2013-2014年黑龙江地区猪流行性腹泻病毒检测及M基因的遗传进化分析
2017-10-23刘佳丽王瑞翀李一经唐丽杰徐义刚乔薪瑗
刘佳丽,王 梓,赵 莉,王瑞翀,李一经,王 丽,唐丽杰,徐义刚,刘 敏,乔薪瑗
(1.东北农业大学动物医学学院 , 黑龙江 哈尔滨 150030 ; 2. 黑龙江省疾病控制中心 , 黑龙江 哈尔滨 150030)
2013-2014年黑龙江地区猪流行性腹泻病毒检测及M基因的遗传进化分析
刘佳丽1,王 梓1,赵 莉1,王瑞翀2,李一经1,王 丽1,唐丽杰1,徐义刚1,刘 敏1,乔薪瑗1
(1.东北农业大学动物医学学院 , 黑龙江 哈尔滨 150030 ; 2. 黑龙江省疾病控制中心 , 黑龙江 哈尔滨 150030)
猪流行性腹泻是由猪流行性腹泻病毒(PEDV)引起的一种严重的病毒性传染病。本试验于2013-2014年在黑龙江省6个地级市的10个规模化猪场采集新生哺乳仔猪临床表现为腹泻症状的粪便与小肠,进行RT-PCR检测,表明PEDV检出率为56% ,PEDV+TGEV检出率为6%,PRV检出率为10%,TGEV检出率为10%,结果表明,引起黑龙江省规模化猪场新生仔猪腹泻的主要病原是PEDV,同时也存在与其他腹泻病毒的混合感染。针对阳性病料扩增出的6株PEDV的M基因序列进行遗传进化分析,6株PEDV-M基因序列之间的核苷酸同源性为98.2%~99%,与疫苗株CV777的核苷酸同源性为97.6%~98.2%。利用MEGA 6.0构建遗传进化树,结果显示,扩增出的6条PEDV-M基因的遗传进化树分为两大系,大部分遗传距离较近,处于同一分支。其中LJJHD-M-13和LJJQ-M-14与泰国KU06RB08、泰国KU07RB08的亲缘关系较近,LJJC-M-14、LJJJ-M-14、LJJH-M-13、LJMM-M-14这4株毒株与韩国M1595、韩国CPF259的亲缘关系较近。同时扩增出的6条PEDV-M基因与CV777毒株非一系,与其亲缘关系较远,说明这些地区的PEDV流行毒株已经发生变异,形成了独特的流行进化分支。因此,本试验通过对变异毒株基因序列进行分析,以期为研制新的疫苗和预防控制该病提供实验数据和理论基础。
猪流行性腹泻病毒 ; M基因 ; 序列分析 ; 遗传进化分析
猪流行性腹泻(Porcine epidemic diarrhea,PED)是由猪流行性腹泻病毒引起的,以严重腹泻及致死性水样腹泻为特征的猪肠道传染病。感染的病猪死亡率可达到50%~90%[1],给我国养猪业造成巨大的经济损失。
PEDV基因组中S、E、M和N基因分别编码纤突(S)蛋白、包膜(E)蛋白、膜(M)蛋白、核衣壳(N)蛋白。M基因的核苷酸序列与其他的冠状病毒同源性较差(约50%),但在不同的PEDV毒株中高度保守[2]。为探究黑龙江地区规模化养猪场新生仔猪PEDV的发病率以及与该病流行相关的潜在风险因素,本试验对黑龙江省的规模化猪场新生仔猪腹泻病料进行了RT-PCR检测和PEDV-M基因遗传进化分析,为黑龙江省地区规模化养猪场预防和控制猪流行性腹泻提供实验数据和理论基础。
1 材料与方法
1.1 样品的采集 本试验于2013-2014年间,从黑龙江省地区规模化养猪场采集125份新生仔猪腹泻病料,其中哈尔滨市的道外区猪场15份,哈尔滨市的巴彦县洼兴镇猪场15份,鸡西市城子河区猪场10份,鸡西市鸡冠区猪场10份,鸡西市恒山区猪场10份,牡丹江市穆棱区猪场15份,鹤岗市兴安区猪场10份,鹤岗市工农区猪场10份,佳木斯市前进区猪场20份,齐齐哈尔市甘南县猪场10份。并参照文献[3-7]的方法采集样品。
1.2 样品的处理 将粪便样品称重,按1∶1比例加入粪便裂解液,涡旋震荡后4 ℃ 梯度离心,反复冻融后于-70 ℃保存。
1.3 病毒RNA的提取及反转录 按照TRIZol 法提取病毒RNA。以病料总RNA为模板进行反转录, -20 ℃保存备用。按照文献[8-10]中的方法进行PEDV-M基因的克隆,并将阳性重组质粒送至上海生工生物工程技术服务有限公司测序。
1.4 RT-PCR检测 分别以PEDV、TGEV和PRV反转录产物为模板,同时设置阴性对照,用rTaq 酶进行RT-PCR检测。
1.5 PEDV-M基因的遗传进化分析 利用DNAMAN软件,将6条PEDV-M基因的核苷酸序列与GenBank中已发表的19个参考毒株(见表1)的M基因的核苷酸序列进行同源性比较,并且利用MEGA 6.0绘制进化树,对PEDV-M基因的遗传进化进行分析。
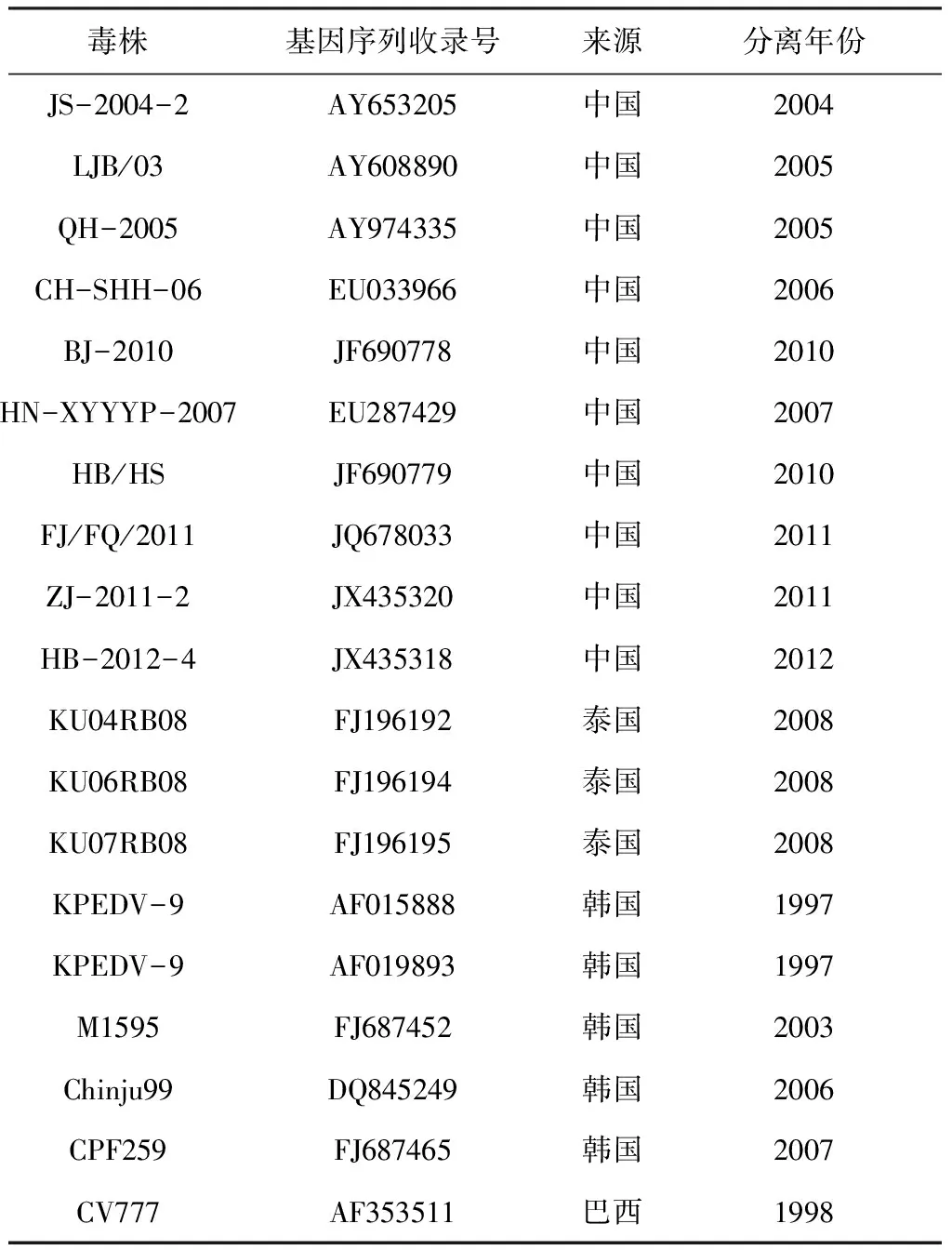
表1 参考毒株序列
2 结果
2.1 腹泻病料检测结果 RT-PCR方法检测结果见表2,PEDV检出率为56%,PEDV+TGEV检出率为6%,PRV检出率为10%,TGEV检出率为10%。PEDV的发生均在2-10日龄的新生哺乳仔猪阶段,一年四季均可发生,病死率达100%。
2.2 PEDV-M基因的扩增及遗传进化分析 6条PEDV- M基因核苷酸同源性为98.2%~99%,与疫苗株CV777的核苷酸同源性为97.6%~98.2%。将6条PEDV-M基因与GenBank中已经发表的19株PEDV毒株的M基因进行比对,并利用MEGA 6.0构建遗传进化树,由图1可以看出: 6条PEDV-M基因的遗传进化树分为二个大系,大部分遗传距离较近,处于同一分支。其中LJJHD-M-13和LJJQ-M-14与泰国KU06RB08、泰国KU07RB08的亲缘关系较近,LJJC-M-14、LJJJ-M-14、LJJH-M-13、LJMM-M-14这4株毒株与韩国M1595、韩国CPF259的亲缘关系较近,同时扩增出的6条PEDV-M基因与CV777毒株非一系,与其亲缘性较远。
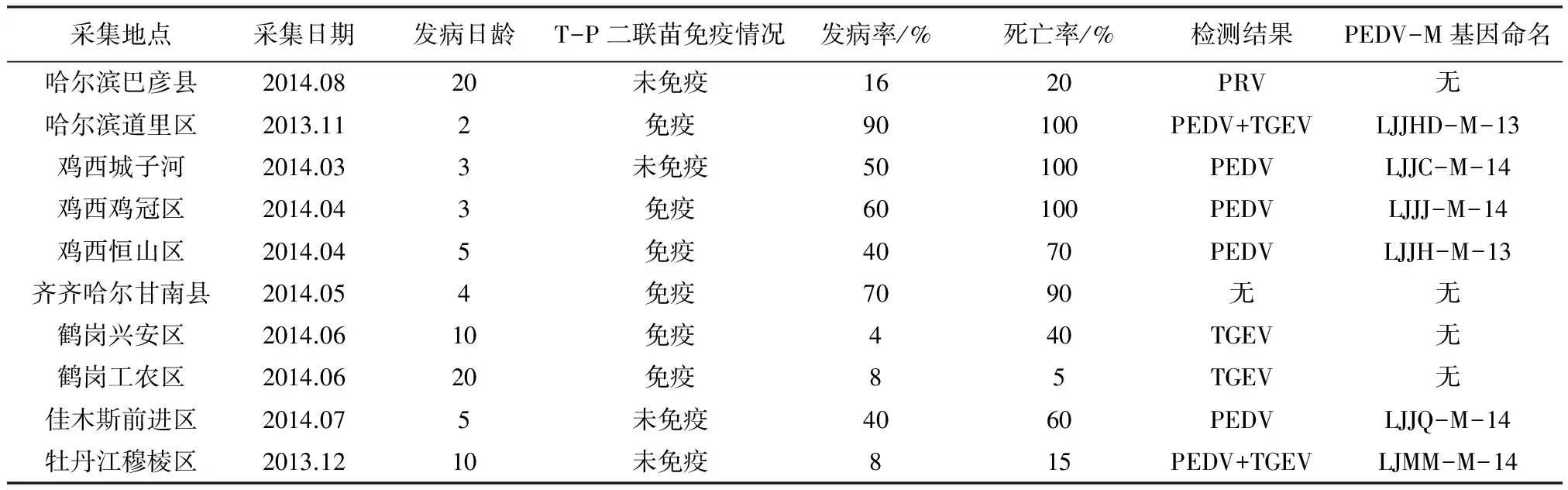
表2 黑龙江省规模化猪场病原检测结果

图1 PEDV -M基因遗传进化树
3 讨论
本研究对2013-2014年黑龙江省的哈尔滨市、鸡西市、牡丹江市、鹤岗市、佳木斯市和齐齐哈尔市6个地区发生新生仔猪腹泻的10个规模化养猪场进行采样,共收集病料125份。在本次调查中,母猪经过T-P二联疫苗免疫后新生仔猪仍可感染PEDV,由此说明疫苗免疫或者母源抗体并不足于保证新生仔猪免受现有PEDV流行毒株的感染。通过PCR扩增得到的PEDV-M基因核苷酸同源性比较发现,获得的6条M 基因序列同源性较高,且与疫苗毒株CV777 M基因的同源性也比较高,说明M基因比较保守,是研究PEDV遗传演化的优先选择基因。由PEDV-M基因的遗传进化分析可知,扩增出的这6条PEDV-M基因与CV777非一系,亲缘关系较远,与泰国毒株、韩国毒株为一系,亲缘关系较近,表明这些地区的 PEDV流行毒株已经发生变异,形成了独特的流行进化分支。通过对变异毒株基因序列的遗传进化分析,可为新疫苗和免疫方法的研究奠定基础,对预防和控制该病具有重要的意义。
[1] Sun D, Wang X, Wei S,etal. Epidemiology and vaccine of porcine epidemic diarrhea virus in China: a mini-review[J]. Journal of Veterinary Medical Science, 2016, 78(3):355-363.
[2] Chen J, Liu X, Shi D,etal. Complete Genome Sequence of a Porcine Epidemic Diarrhea Virus Variant[J]. Journal of Virology, 2012, 86(6):3 408.
[3] Huang M Z, Wang H, Wang S Y,etal. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus samples obtained from farms in Gansu, China [J]. Genetics & Molecular Research Gmr, 2015, 15(1).
[4] 李长龙, 陈建飞, 张鑫,等. 猪流行性腹泻病毒野毒株/疫苗株RT-PCR鉴别诊断方法的建立[J]. 中国兽医杂志, 2014, 50(6):6-8.
[5] Zhao P, Tan C, Dong Y,etal. Genetic variation analyses of porcine epidemic diarrhea virus isolated in mid-eastern China from 2011 to 2013 [J]. Canadian journal of veterinary research = Revue canadienne de recherche veterinaire, 2015, 79(1):8.
[6] Kim S H, Lee J M, Jung J,etal. Genetic characterization of porcine epidemic diarrhea virus in Korea from 1998 to 2013 [J]. Archives of Virology, 2015, 160(4):1 055-1 064.
[7] Zhao Z P, Yang Z, Lin W D,etal. The rate of co-infection for piglet diarrhea viruses in China and the genetic characterization of porcine epidemic diarrhea virus and porcine kobuvirus[J]. Acta Virologica, 2015, 60(1):55.
[8] Gao Y, Kou Q, Ge X,etal. Phylogenetic analysis of porcine epidemic diarrhea virus field strains prevailing recently in China [J]. Archives of Virology, 2013, 158(3):711-715.
[9] Kang J H, Kwon D H, Chung T W,etal. Development of a simple and rapid immunochromatographic strip test for diarrhea-causative porcine rotavirus in swine stool [J]. Journal of Virological Methods, 2007, 146(1-2):74-79.
[10] Lorenzetti E, Stipp D T, Possatti F,etal. Diarrhea outbreaks in suckling piglets due to rotavirus group C single and mixed (rotavirus groups A and B) infections[J]. Pesquisa Veterinária Brasileira, 2014, 34(5):391-397.
Molecularcharacterizationandphylogeneticanalysisofporcineepidemicdiarrheavirus(PEDV)fieldisolatesinHeilongjiang,China,2013—2014
LIU Jia-li1, WANG Zi1, ZHAO Li1, WANG Rui-chong2, LI Yi-jing1, WANG Li1, TANG Li-jie1, XU Yi-gang1, LIU Min1, QIAO Xin-yuan1
(1. Northeast Agricultural University, College of Veterinary Medicine, Harbin 150030, China; 2. Heilongjiang Provincial Center for Disease Control, Harbin 150030, China)
Porcine epidemic diarrhea virus (PEDV) is the etiological agent of Pig epidemic diarrhea (PED).In this study, feces and small intestine of the neonatal piglets that were noted with signs of diarrhea were collected form 10 large-scale pig farms in six cities of Heilongjiang province from 2013 to 2014, and the RT-PCR analysis was used to detected the pathogens. The results showed that 56% of the samples were positive for PEDV, 6% of the samples were positive for PEDV and TGEV, 10% of the samples were positive for PRV and 10% of the samples were positive for TGEV. Our findings indicated that PEDV was the main pathogen that caused diarrhea of neonatal piglets in Heilongjiang province, and there was also a mixed infection with other diarrhea viruses. There were six PEDV-M gene sequences were detected by RT-PCR; The results showed that the nucleotide homology was 98.2%~99% among these genes, and the nucleotide homology between these stains and vaccine strain CV777 was 97.6%~98.2%.Genetic evolutionary tree was built by MEGA 6.0, and the result showed that the 6 PEDV-M genes were divided into two series, and most of the genetic distance was velatively close at the same branch. Compared with other Chinese isolates and foreign isolates, the 6 PEDV isolates had a closer genetic relationship with the Korea isolate M1595, CPF259 and Thailand isolates KU06RB08, KU07RB08, but had more distant genetic relationship with the vaccine strain CV777. Our results indicated that the PEDV epidemic strains in these areas had been mutated to form a unique branch of the popular evolution.
Porcine epidemic diarrhea virus ; M gene ; Sequence analysis ; Genetic evolution analysis
QIAO Xin-yuan
S852.62
A
0529-6005(2017)08-0007-03
2017-02-16
黑龙江省科学基金项目(C2016028)
刘佳丽(1991-),女,硕士,从事预防兽医学研究,E-mail:313225950@qq.com
乔薪瑗,E-mail:qiaoxinyuan@126.com
