高效液相色谱法同时检测植物甾醇与植物甾醇酯
2017-07-19杨福明
杨福明 马 莺
(哈尔滨工业大学食品科学与工程学院1,哈尔滨 150090) (九三粮油工业集团有限公司2,哈尔滨 150060)
高效液相色谱法同时检测植物甾醇与植物甾醇酯
杨福明1,2马 莺1
(哈尔滨工业大学食品科学与工程学院1,哈尔滨 150090) (九三粮油工业集团有限公司2,哈尔滨 150060)
采用反相高效液相色谱,对同时检测植物甾醇和甾醇酯的分析方法进行了研究。通过检测条件优化,植物甾醇和甾醇酯的12种不同组分在以丙酮和乙腈(3∶1)为流动相,柱温30 ℃,流动相流速1.0 mL/min,等度洗脱的条件下,达到最好分离效果。此方法不需要样品的前处理,分析时间25 min,检出限为5.1~1.1 μg/mL,定量限为17.0~137.0 μg/mL,回收率93.81%~111.98%。植物甾醇与甾醇乙酸酯在0.01~1.0 mg/mL的浓度范围内呈线性相关,甾醇亚油酸酯与甾醇油酸酯在0.1~1.0 mg/mL范围内具有良好的线性关系,标准曲线相关系数均超过0.998。该方法可应用于多种甾醇和甾醇酯产品的同时检测。
植物甾醇 植物甾醇酯 高效液相色谱法 同时检测
植物甾醇是一种天然活性物质,主要由菜油甾醇、豆甾醇、β-谷甾醇几种主要成分组成,具有降低胆固醇[1-2]、抗癌[3]、预防心脑血管疾病[4]等十分重要的生理功能,在食品、保健品、化妆品、药品等领域有着广泛应用。植物甾醇本身的理化性质决定了其不溶于水,在油中的溶解度小,降低了实际的使用价值。为扩大甾醇的应用范围,当前普遍采用的方法是将植物甾醇进行酯化,得到甾醇乙酸酯、甾醇亚油酸酯、甾醇油酸酯等甾醇酯产品[5-8]。植物甾醇酯有更优的脂亲和性,在油中的溶解度可以大大提高,扩大了甾醇的应用范围。
测定植物甾醇的方法很多,有紫外分光光度法、液相色谱法、气相色谱法、气相色谱-质谱法[9-12]。检测植物甾醇酯的研究并不多见,目前仅有关于液相色谱检测甾醇烟酸酯[13]、气相色谱检测甾醇亚麻酸酯[14]的报道。液相法检测甾醇烟酸酯采用的是以纯甲醇为流动相,此流动相如果用来检测甾醇乙酸酯需要35~40 min,且无法检测出甾醇亚油酸酯和甾醇油酸酯。气相色谱法检测甾醇亚麻酸酯采用的是先将甾醇酯皂化预处理,变成游离甾醇,通过内标法检测甾醇的含量然后折算成甾醇酯的含量,气相色谱法前处理过程较复杂,检测温度高。普通液相色谱法同时检测多种甾醇和甾醇酯的研究鲜见报道,因此,采用液相色谱的方法在较短时间内同时分析多种植物甾醇和甾醇酯的含量,对促进甾醇酯产品质量指标分析方法的应用很有意义。
本研究以大豆甾醇、大豆甾醇乙酸酯、大豆甾醇亚油酸酯、大豆甾醇油酸酯含量的检测为研究对象,采用反相液相色谱法,通过优化不同的流动相、流速、等度洗脱、梯度洗脱、色谱柱、柱温等条件,建立在较短时间内同时分析12种植物甾醇和甾醇酯组分的检测方法。
1 材料与方法
1.1 材料与仪器
大豆甾醇(≥95%)、大豆甾醇乙酸酯(≥95%)、大豆甾醇亚油酸酯(≥60%)、大豆甾醇油酸酯(≥85%):九三粮油工业集团有限公司;菜油甾醇标准品(≥98%)、豆甾醇标准品(≥98%)、β-谷甾醇标准品(≥98%):成都普瑞法科技开发有限公司;油酰氯(≥99%)、亚油酸(≥99%):Sigma公司;乙酸酐、NaCl、NaHCO3、正己烷:天津科密欧试剂公司;色谱纯丙酮、色谱纯乙腈:霍尼韦尔公司色谱纯试剂。Waters 2695液相色谱仪、UV2489检测器:美国沃特世公司。
1.2 植物甾醇酯标准品的制备
甾醇乙酸酯、甾醇亚油酸酯、甾醇油酸酯均为实验室合成。为提高合成效率,甾醇油酸酯与亚油酸酯的合成采用酰氯法。但由于没有商业化的亚油酰氯,因此在参考前人方法的基础上[13],以高纯度的亚油酸(≥99%)为原料,实验室合成了高纯度亚油酰氯,用于甾醇亚油酸酯标准品的合成。具体合成步骤为:取3个50 mL磨口三角瓶,分别加入菜油甾醇、豆甾醇、β-谷甾醇标准品各100 mg,然后加入摩尔比1∶1.1的乙酸酐(油酰氯或亚油酰氯),连接与磨口三角瓶相配套的冷凝管,135 ℃(油酰氯与亚油酰氯反应温度为85 ℃)加热条件下,冷凝回流反应1 h。冷却后往三角瓶中加入过量的饱和NaHCO3溶液,漩涡振荡,充分混合,除掉未反应的酸性物质。加入5 mL正己烷,磨口三角瓶加塞颠倒混匀数次,萃取合成的甾醇酯产物。加入饱和的NaCl溶液使液面升至瓶口处,吸取上层有机相,氮气吹干。最后取少量产物液相色谱上机检测,采用面积归一化法和标准品验证法对合成的标准品进行定量分析,产物中几乎无未反应的甾醇时(甾醇酯质量分数≥98%),则认为得到了合格的甾醇酯标准品。
1.3 植物甾醇与甾醇酯标准溶液的配制
准确称量菜油甾醇、豆甾醇、β-谷甾醇标准品各50 mg溶于50 mL丙酮中,制成这几种物质的混合标准溶液。精确量取0.1、0.5、2.5、5、10 mL的混合标准溶液,用丙酮定容至10 mL,制成菜油甾醇(0.01~1.0 mg/mL)、豆甾醇(0.01~1.0 mg/mL)、β-谷甾醇(0.01~1.0 mg/mL)5个不同浓度的标准溶液。每个浓度的标准溶液在液相色谱仪自动进样器上吸取10 μL进行含量检测,制成5点标准曲线。甾醇乙酸酯标准曲线的制备与植物甾醇相同,甾醇亚油酸酯、甾醇油酸酯标准曲线的浓度稍有不同,各组分质量浓度范围为0.1~1.0 mg/mL。
1.4 样品的制备与色谱条件
称取适量的大豆甾醇、甾醇乙酸酯、甾醇亚油酸酯、甾醇油酸酯样品用合适体积的丙酮定容,取其中的1.5 mL用0.45 μm滤垫过滤后置于1.5 mL进样瓶中备用。
色谱柱采用C18反相色谱柱(4.6 mm×250 mm×5 μm),流动相流速1.0 mL/min,进样量10 μL,柱温30 ℃,紫外检测器波长210 nm。
2 结果与讨论
2.1 流动相的确定
采用传统的甲醇、乙腈为流动相,对甾醇和甾醇酯产品进行了检测,发现这2种流动相可以检测出甾醇和甾醇乙酸酯的各种组分,甾醇乙酸酯的检测时间过长,达到了40 min。但以甲醇、乙腈为流动相时,未能检测出甾醇亚油酸酯和甾醇油酸酯。经过其他流动相的试验和探索,在参考安广杰等[15]测定甘三酯组成研究的基础上略作改进,得到了最好的分离效果。安广杰等[15]采用的流动相为乙腈+丙酮(36.4∶63.6),本研究通过梯度洗脱来优化乙腈与丙酮最佳比例,流动相确定为乙腈+丙酮(1∶3)。这样的流动相比例可能与甾醇、甾醇酯的极性特点有关,根据相似相溶原理,甲醇、乙腈的极性太强,不适合甾醇油酸酯和甾醇亚油酸酯的分析。
2.2 色谱柱柱温的选择
流动相确定后,为使植物甾醇和植物甾醇酯各组分达到最佳的分离度,对25~45 ℃柱温时的检测结果进行了考查。25 ℃时色谱峰的峰宽较大,但检测时间长,45 ℃时色谱峰的峰宽较小,峰值高,但不同组分的分离效果不好。30 ℃时色谱峰的峰形最好,且检测时间适中,因此选择色谱柱温度为30 ℃。此时大豆甾醇、甾醇乙酸酯、甾醇亚油酸酯、甾醇油酸酯标准品的检测结果如图1所示。从色谱图可以看出,在此检测条件下色谱的基线平稳,25 min的分析时间内植物甾醇和甾醇酯的12种不同组分得到了较好的分离。这种现象的产生可能是由于,在其他条件都不变的情况下,柱温不同峰高和峰宽会随之改变。柱温高有利于传质,柱箱温度越高,峰高越高,峰宽越窄,但是峰与峰之间的间距会越小。柱温过高时,分配系数变小,不利于分离。反之柱温越低,峰高越低,峰宽越宽,峰之间的间距越大,有利于组分的分离。但温度过低,被测组分传质阻力增加,使色谱峰扩张,甚至拖尾,所以不一定温度越低分离越好。试验通过选择最佳柱温,使被检测组分既完全分离,又不使峰形扩展、拖尾,达到最合适的效果。
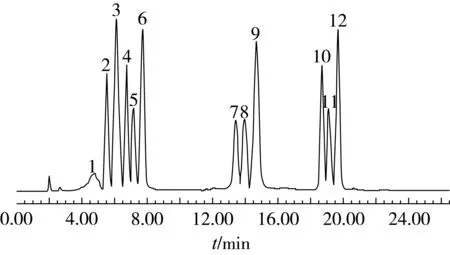
注:色谱峰1~12分别为菜油甾醇、豆甾醇、β-谷甾醇、菜油甾醇乙酸酯、豆甾醇乙酸酯、β-谷甾醇乙酸酯、菜油甾醇亚油酸酯、豆甾醇亚油酸酯、β-谷甾醇亚酸酯、菜油甾醇油酸酯、豆甾醇油酸酯、β-谷甾醇油酸酯。图1 30 ℃条件下甾醇与甾醇酯标准品的液相色谱图
2.3 甾醇酯标准品的确认
甾醇酯标准品合成反应中采取了酸过量的方式,反应完成后对合成产物使用过量的饱和NaHCO3溶液进行处理,去除未反应的酸性物质。
标准品合成后采用2种方式对其真实含量进行确认。面积归一化法:由于在试验得到的最佳检测条件下,甾醇与甾醇酯各组分全部流出色谱柱,可以实现很好的分离,且基线平稳,在色谱图上都显示色谱峰,因此可以采用面积归一化法从10、20、40 μL 3个不同的进样量对合成的甾醇酯标准品纯度进行确认。标准物质验证法:合成反应后剩余甾醇的量很少,用甾醇标准曲线对未反应的甾醇定量,每个样品重复检测3次,然后根据未反应甾醇的量计算出甾醇酯含量。2种确认方式得到的甾醇酯标准品含量如表1所示,得到结果的一致性较好,终含量取两者平均值。
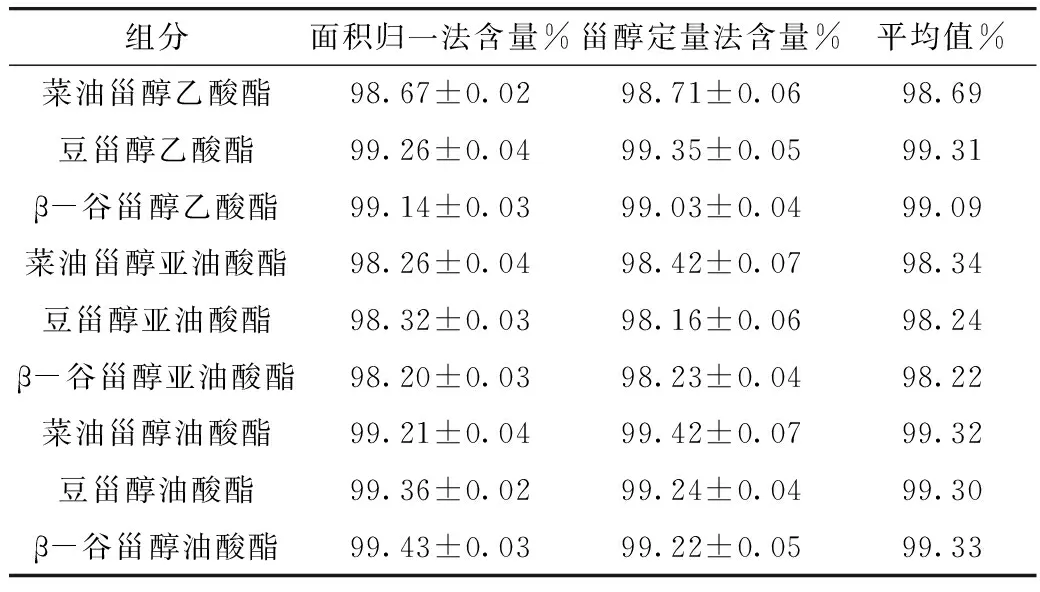
表1 甾醇酯标准品含量的确认结果
2.4 检出限、定量限与精确度
检出限是指在适当的置信概率被检出的组分最低浓度或最小量,一般表示为分析物质的浓度。本试验中检出限和定量限通过以下方法获得:对接近标准曲线最低浓度的甾醇和甾醇酯的标准溶液重复检测10次,计算得出平均值和标准偏差。标准偏差值的3倍即为检出限,标准偏差值的10倍为定量限。12种不同组分的检测和计算结果如表2所示,从表2可以看出不同的甾醇与甾醇酯组分的检出限为5.1~41.1 μg/mL,定量限为17.0~137.0 μg/mL。从各种组分检测含量的平均值也可以看出实测值与实际含量相差不大,方法的准确度好,达到了GB/T 27404—2008《实验室质量控制规范 食品理化检测》中检测方法确认的技术要求。另外,从表2中可以看出SD值普遍较小,RSD值为4.37%~8.98%,方法的精确度较高。
2.5 线性范围与回收率
大豆甾醇、甾醇乙酸酯标准品在质量浓度0.01~1.0 mg/mL范围内作标准曲线,甾醇亚油酸酯、甾醇油酸酯标准品在质量浓度0.1~1.0 mg/mL范围内制作标准曲线,每种组分标准曲线的相关系数都超过了0.998,线性关系较好,浓度范围和相关系数符合GB/T 27404—2008。
对各组分的具体含量进行了加标回收试验,取4份已知浓度的样品溶液,其中3份加入分别加入高、中、低不同量的标准物质,4份按相同的分析步骤分析,每份样品重复检测3次。加标的1份所得的结果减去未加标1份所得的结果,其差值同加入标准物质的理论值之比即为样品加标回收率。具体的试验结果如表3所示,从试验结果可知甾醇与甾醇酯各组分的加标回收率为93.81%~111.98%,除个别数据(93.81%、111.98%)外,回收率达到GB/T 27404—2008中的要求(95%~105%)。
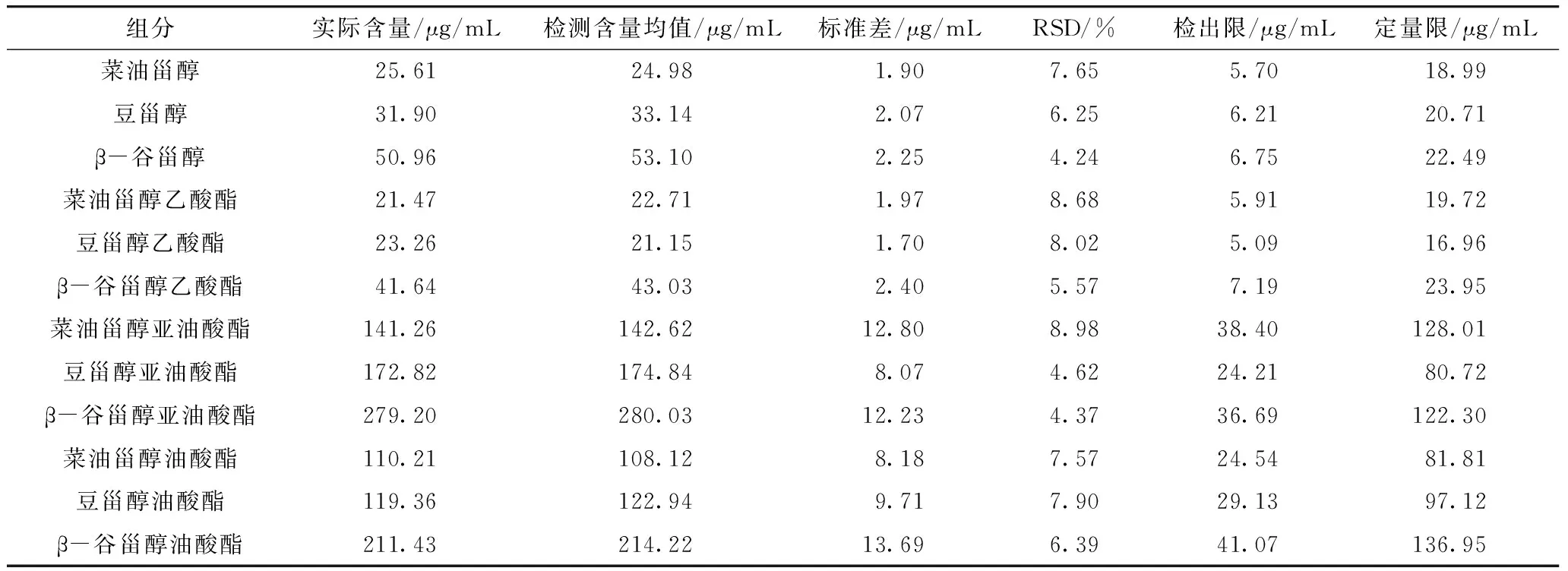
表2 甾醇、甾醇酯各组分的检出限、定量限和精确度
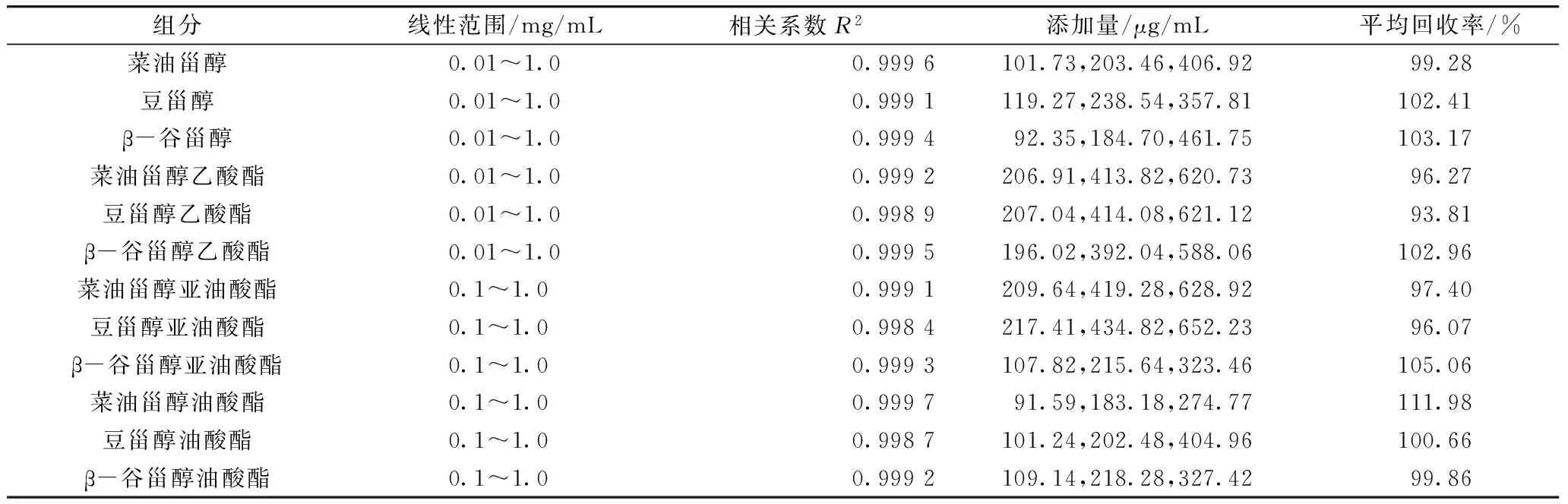
表3 甾醇、甾醇酯各组分的线性范围和加标回收率
2.6 实际样品的定量分析
用标准曲线对市售大豆甾醇(≥95%)、甾醇油酸酯(≥85%)、甾醇乙酸酯(≥95%)、甾醇亚油酸酯(≥60%)几种产品进行定量分析,每个样品重复检测3次,样品检测的色谱图分别如图2~图5所示,各个产品总含量与每种组分的含量的检测结果如表4所示。几种不同规格的产品大豆甾醇、甾醇油酸酯、甾醇乙酸酯、甾醇亚油酸酯的质量分数分别为96.41%、91.53%、95.73%、69.40%,通过实际含量的检测可以看出所测产品的总含量均合格,各种不同的组分也能得到准确的定量。大豆甾醇中含有水分以及微量不溶性杂质,甾醇油酸酯含有未酯化甾醇和少量的其他类型脂肪酸酯,甾醇乙酸酯含有少量的未酯化甾醇,甾醇亚油酸酯是含有较大量的甾醇油酸酯,经试验发现这些产品中的非目标分析物未对分离产生影响。样品中不含有蛋白、盐类等其他杂质,含有的少量不溶性杂质被滤膜过滤,直接进样对色谱柱的影响不大,因此可以采用直接进样的方式。

图2 大豆甾醇产品的检测色谱图
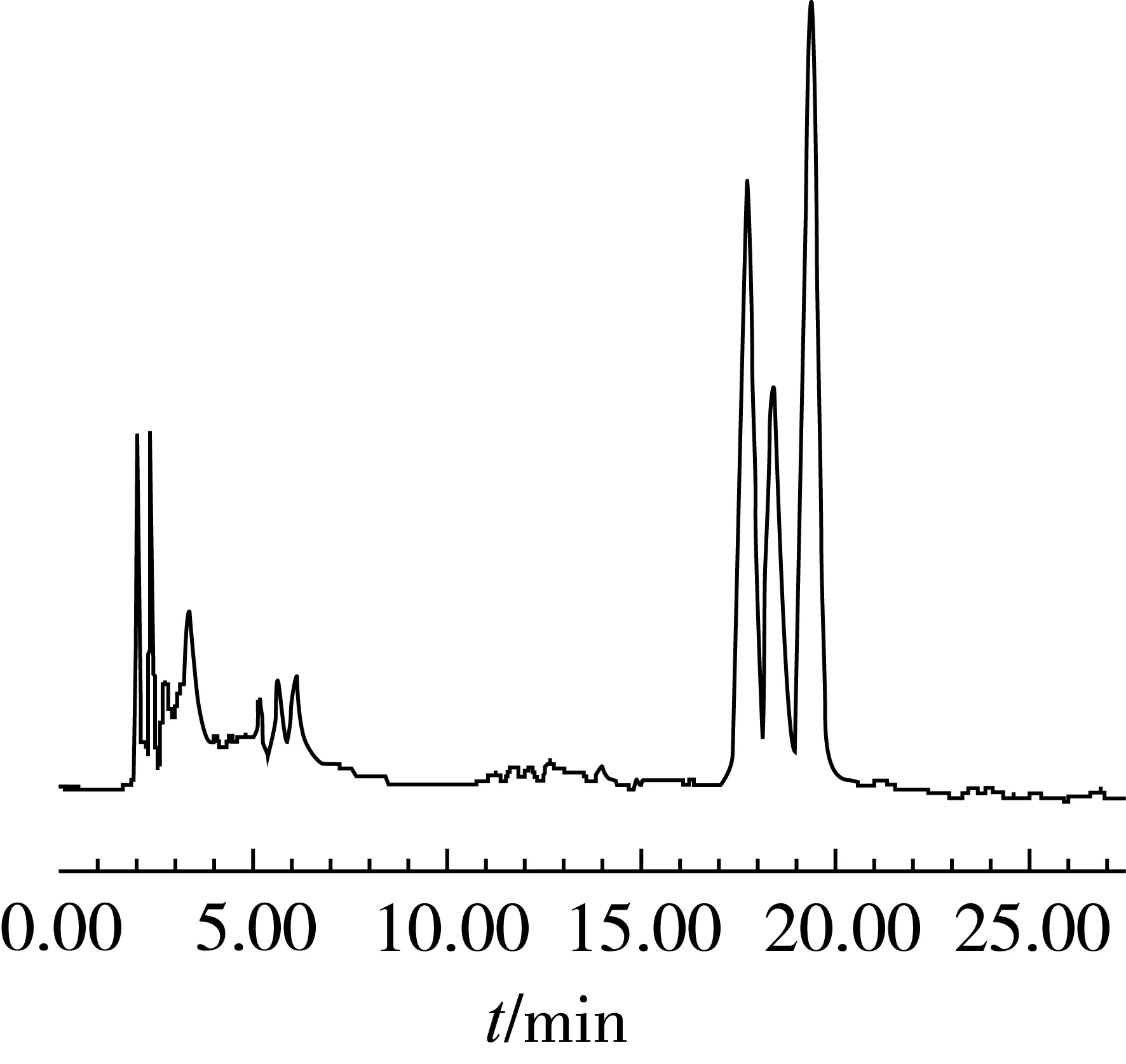
图3 甾醇油酸酯产品的检测色谱图
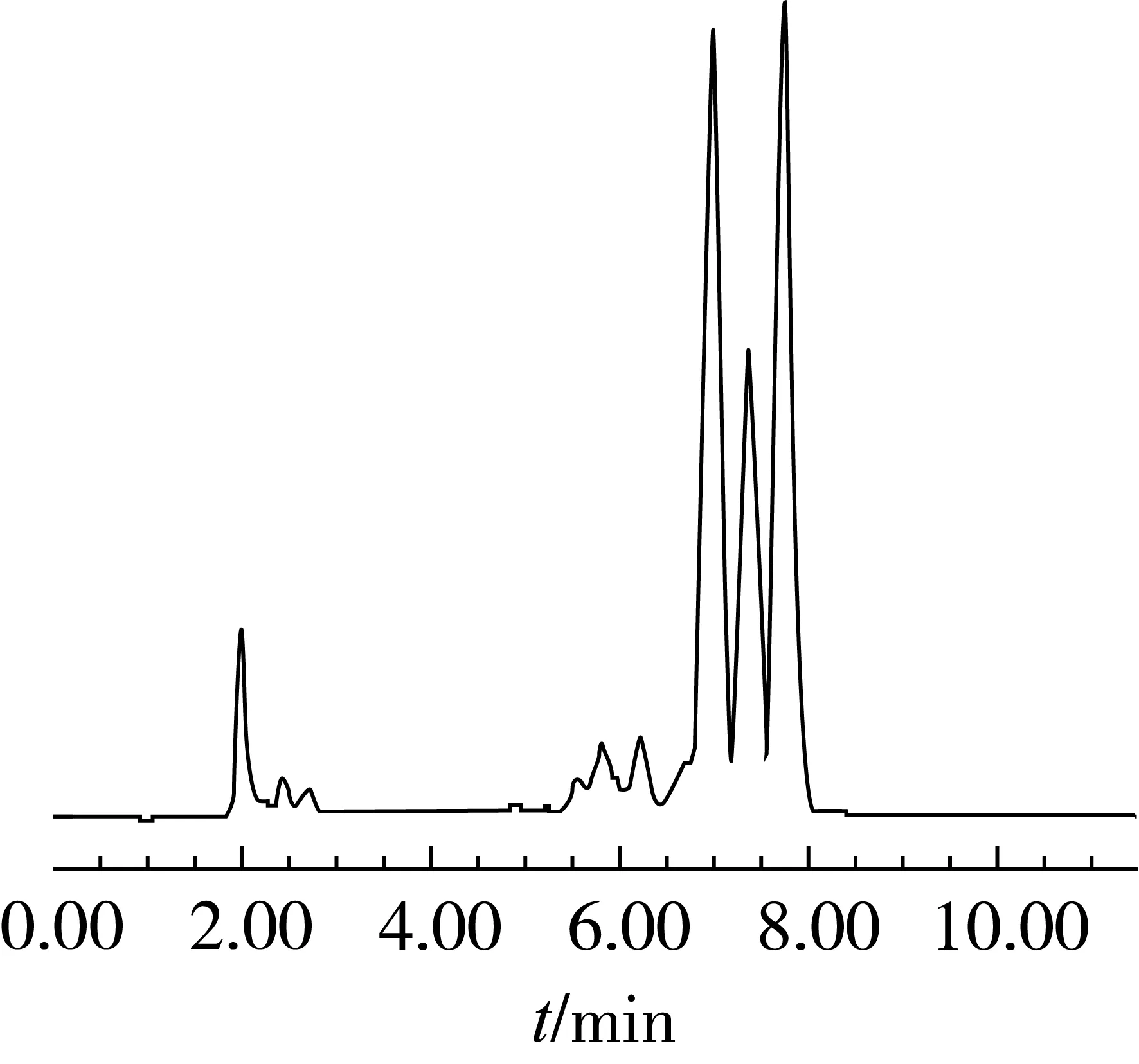
图4 甾醇乙酸酯产品的检测色谱图
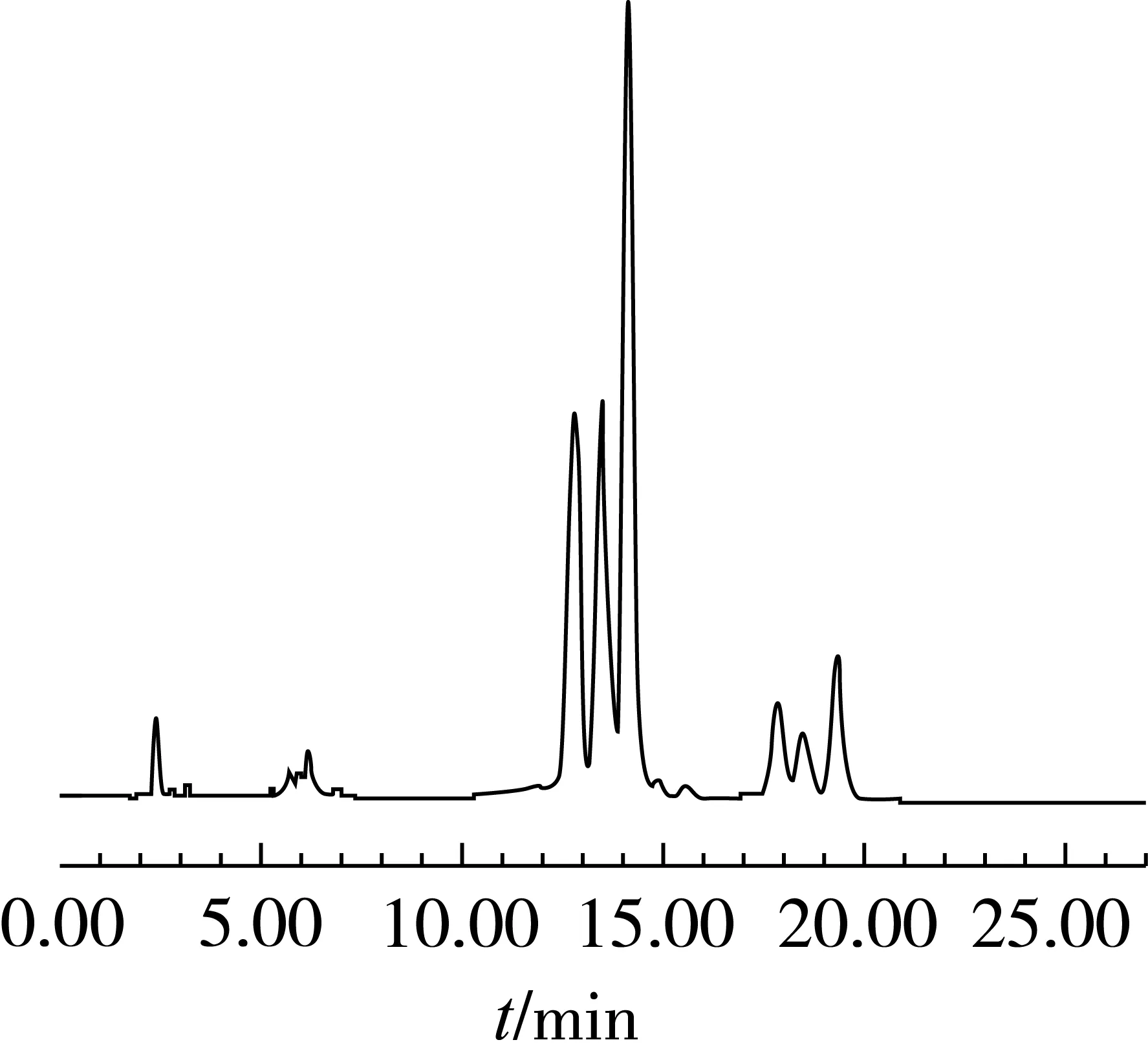
图5 甾醇亚油酸酯产品的检测色谱图
表4 植物甾醇、甾醇酯产品实际含量的检测结果

产品总量/%菜油甾醇或酯/%豆甾醇或酯/%β-谷甾醇或酯/%大豆甾醇96.41±0.5824.39±0.1526.42±0.1645.02±0.27甾醇乙酸酯95.73±0.7924.22±0.2026.23±0.2244.71±0.37甾醇亚油酸酯69.40±1.7117.56±0.4319.02±0.4732.41±0.80甾醇油酸酯91.53±0.6223.16±0.1625.08±0.1742.74±0.29
3 结论
采用普通的液相色谱方法可以在25 min的分析时间内实现12种不同的甾醇和甾醇酯组分的分离,达到对植物甾醇和植物甾醇酯各组分同时检测的目的。研究发现,液相色谱法不需要样品的前处理、检测时间短、定量准确。尽管试验过程中,当样品浓度过高时部分组分的分离度会降低,色谱峰没有完全分离,且由于试验条件所限,本研究未对甾醇亚麻酸酯、甾醇月桂酸酯、甾醇烟酸酯等其他更多形式产品的同时检测进行研究。但是从理论上分析,此方法可以应用在其他类型产品的含量检测上。建议在检测工作中可根据实际情况进行适当的条件优化。
[1]Brufau G,Canela M A,Rafecas M.Phytosterols:physiologic and metabolic aspects related to cholesterol-lowering properties[J].Nutrition Research,2008,28:217-225
[2]Mark M A,Hang J L,Dussault P H.Phytosterol stearate esters elicit similar responses on plasma lipids and cholesterol absorption but different responses on fecal neutral sterol excretion and hepatic free cholesterol in male Syrian hamsters[J].Nutrition Research,2011,31:537-543
[3]Llaverias G,Escolà G J C,Lerma E.Phytosterols inhibit the tumor growth and lipoprotein oxidizability induced by a high-fat diet in mice with inherited breast cancer[J].Journal of Nutritional Biochemistry,2013,24(1):39-48
[4]Earnest C P,Catherine R M,Lemieux I.Examination of encapsulated phytosterol ester supplementation on lipid indices associated with cardiovascular disease[J].Nutrition,2007,23:625-633
[5]陈茂彬,李万芬,吴谋成.植物甾醇乙酸酯的合成研究[J].化学与生物工程,2003,5:33-34 Chen M B,Li W F,Wu M C.Study on synthesis of phytosterols acetate[J].Chemistry & Bioengineering,2003,5:33-34
[6]Valange S,Beauchaud A,Barrault J.Lanthanum oxides for the selective synthesis of phytosterol esters:Correlation between catalytic and acid-base properties[J].Journal of Catalysis,2007,251:113-122
[7]Manuel S R,Barrault J,Valange S.Selective synthesis of phytosterol esters from natural sterols and fatty methyl esters over Mg-containing solid catalysts[J]. Comptes Rendus Chimie,2011,14:656-662
[8]Hellner G,Töke E R,Nagy V,et al.Integrated enzymatic production of specific structured lipid and phytosterol ester compositions[J].Process Biochemistry,2010,45:1245-1250[9]窦巍巍.酯交换大豆油技术及植物甾醇快速检测方法建立[D].沈阳:东北农业大学,2012:18-19 Dou W W.Transesterified soybean oil technology and establishment of rapid determination of phytosterol[D].Shenyang:Northeast Agricultural University,2012:18-19
[10]牟德华,赵玉华,朱艳丽,等.高效液相色谱法测定植物甾醇的研究[J].食品工程,2007,1:62-64 Mou D H,Zhao Y H,Zhu Y L,et al.Determination of phytosterol by HPLC[J].Food Engineering,2007,1:62-64
[11]冯妹元,韩军花,刘成梅,等.常见精练油中植物甾醇测定方法的建立及含量分析[J].中国食品卫生杂志,2006,18(3):197-201 Feng M Y,Han J H,Liu C M,et al.Use of gas chromatography for determination of phytosterols in plant oils[J].Chinese Journal of Food Hygiene,2006,18(3):197-201
[12]徐响,孙丽萍,董捷,等.气相色谱-质谱法测定蜂王浆中的植物甾醇组成[J].食品科学,2010,31(18):317-320 Xu X,Sun L P,Dong J,et al.Sterol composition analysis of royal jelly by gas chromatography coupled with mass spectrometry[J].Food Science,2010,31(18):317-320
[13]施光宗,洪挺,陆豫.植物甾醇烟酸酯的合成及HPLC 法的测定[J].南昌大学学报:理科版,2011,35(4):357-359 Shi G Z,Hong T,Lu Y.Synthesis of bionic ester of phytosterol and anaIysis of it by HPLC[J].Journal of Nanchang University:Natural Science,2011,35(4):357-359
[14]刘昌盛,王明霞,李江涛, 等.α-亚麻酸甾醇酯检测技术研究[J].中国油料作物学报,2009,31(2):239-242 Liu C S,Wang M X,Li J T,et al.Analysis of phytosterol esters of α-linolenic acid[J].Chinese Journal of Oil Crop Sciences,2009,31(2):239-242
[15]安广杰,侯冰冰,王瑛瑶,等.超高效液相色谱法测定油脂中甘三酯组成[J].中国油脂,2011,36(5):55-57. An G J,Hou B B,Wang Y Y,et al.Determination of triacylglycerol composition in oil by ultra performance liquid chromatography[J].China Oils and Fats,2011,36(5):55-57.
Simultaneous Detection of Phytosterols and Phytosterol Esters with HPLC
Yang Fuming1,2Ma Ying1
(College of food science and engineering,Harbin Institute of Technology1,Haerbin 150090) (Jiusan Grains & Oils Industry Group Co.,Ltd2.,Haerbin 150060)
Simultaneous detection method of phytosterols and phytosterol esters was studied by reversed-phase HPLC.Acetone and acetonitrile(3∶1)was used as mobile phase,12 compounds of phytosterols and phytosterol esters could be separated completely under the conditions of flow rate 1.0 mL/min,column temperature 30 ℃ and isocratic elution.Analysis time was only 25 minutes in this method,and there was no sample pretreatment.Limit of detection ranged from 5.1 to 41.1 μg/mL,and limit of quantitative ranged from 17.0 to 137.0 μg/mL,and recovery rates ranged from 93.81% to 111.98%.Sterol linoleate and sterol oleate in the concentration range of 0.01~1.0 mg/mL showed a linear correlation.Linearity range of sterol linoleate and sterol oleate was 0.1~1.0 mg/mL.All the regression coefficients of standard curves were over 0.998.This method could be used in simultaneous detection of different sterol and sterol ester products,and worthy of promotion.
phytosterol,phytosterol esters,HPLC,simultaneous detection
2015-11-13
杨福明,男,1981年出生,博士,食品科学与工程
马莺,女,1961年出生,教授,食品科学与工程
O656.3
A
1003-0174(2017)06-0134-06
