铌对单晶γ-TiAl合金裂纹扩展影响的分子动力学模拟
2017-06-21芮执元罗德春剡昌锋
杨 利,芮执元,罗德春,剡昌锋
(1. 兰州理工大学 机电工程学院,甘肃 兰州 730050;2. 兰州理工大学数字制造技术与应用省部共建教育部重点实验室,甘肃 兰州 730050)
铌对单晶γ-TiAl合金裂纹扩展影响的分子动力学模拟
杨 利1,2,芮执元1,2,罗德春1,2,剡昌锋1,2
(1. 兰州理工大学 机电工程学院,甘肃 兰州 730050;2. 兰州理工大学数字制造技术与应用省部共建教育部重点实验室,甘肃 兰州 730050)
为了研究微观尺度下铌元素对单晶γ-TiAl裂纹扩展过程的影响,运用分子动力学方法,建立γ-TiAl合金的晶体结构模型,模拟边缘裂纹扩展的过程,得到了裂纹扩展的轨迹图和能量演变图,分析了铌元素对γ-TiAl能量和应力-应变关系的影响,进而揭示了铌元素对裂纹扩展的影响。研究结果表明:加入铌元素后,在相同的应变率条件下,试件的断裂时间延长,能量变化曲线有两个波峰并且出现明显的上下波动的现象;随着应变增加,应力先增大后逐渐减小;裂纹扩展缓慢,形成的断面不平滑,而且裂纹扩展的形态也发生变化。
铌; γ-TiAl合金; 分子动力学; 裂纹扩展; 能量
1 引 言
目前普通钛铝合金的主要缺点是室温塑性和断裂韧性较低,以及800℃以上抗高温蠕变和抗高温氧化性能较差[1-2]。高铌钛铝合金以其优越的高温强度和高温抗氧化性能已成为开发耐高温合金的重要发展方向[2]。加入铌(Nb)元素已经成为TiAl合金中提高抗氧化性非常有效的技术手段之一,γ-TiAl合金的成分也从最初的Ti-48Al-2Cr-2Nb逐渐发展到目前所用的Ti-45Al-(5~8)Nb[3]。要使γ-TiAl合金能够合理地被运用到实际工况中,必须了解Nb元素对其结构和性能之间的关系。由于在制造和使用过程中不可避免地会出现微裂纹、空洞等缺陷,而这些缺陷的演化将导致材料的失效和破坏,因此,研究Nb元素的加入对γ-TiAl合金微观结构和缺陷的影响至关重要。分子动力学就是一种计算机模拟实验的方法,是介于真实实验研究与理论计算间的一种方法,是沟通宏观特性与微观信息的桥梁,因而成为分子、原子尺度上研究微观特性的有力工具。付蓉[4]等用分子动力学的方法研究了在恒加载速度下温度对γ-TiAl合金裂纹扩展的影响,结果显示:室温时,裂纹呈微解理扩展,中高温时,裂纹在扩展过程中有位错的发射,裂尖出现钝化现象并且裂纹扩展方向出现偏转;随着温度的升高,裂纹扩展从脆性解理转变为韧性扩展,裂纹扩展速率明显减小,材料塑性增强。蒋孟玲等[5]通过实验方法研究了铌含量对TiAl合金铸态组织的影响,Nb含量为1%时,TiAl-Nb合金铸锭组织主要为单相的γ组织;随Nb含量升高,合金组织主要为α2/γ层片组织;并在层片组织间存在2种偏析,分别是网状β相和γ相,合金的层片晶团平均尺寸逐渐增加,β相的体积分数逐渐升高。余龙[6]等研究了Ti-45Al-8Nb-0.2W-0.2B-0.1Y合金在750和800℃时短时蠕变行为,并进行了扫描电镜原位观察,结果表明:在750℃蠕变时,合金具有稳定的蠕变特征,随应力提高,稳态蠕变阶段变短,其蠕变应力指数为7.5;在800℃蠕变时,低应力下具有明显的稳态蠕变阶段,其蠕变应力指数为4.0,而高应力下几乎没有稳态蠕变,直接进入加速蠕变。Xu[7]等用分子动力学研究了TiAl金属化合物在位错反应中点缺陷的形成过程,通过分子动力学模拟显示在剪切变形中,单一滑移系的运动即能促使空位、位错线及位错环的形成,且间隙原子显示出较强的运动能力,点缺陷与位错的反应程度很大程度取决于缺陷的性质以及滑移面之间的距离,此外,模拟中也显示出位错反应是许多点缺陷生成的来源。Tang[8]等人用分子动力学模拟了γ-TiAl单晶的空洞开裂过程,结果表明:位错核的连续产生和剪切循环的扩展使得空洞开裂,初始屈服强度随着试件尺寸和空洞体积分数的增加而减小,随着应变率的增加而增加。曲洪磊[9]等人用分子动力学方法对γ-TiAl单晶纳米杆在室温下的拉伸变形过程进行了模拟研究,结果表明γ-TiAl单晶纳米杆在室温下的塑性变形机制为孪生和普通位错,进入塑性变形后单晶纳米杆中开始出现层错等缺陷,变形过程中积累的应变能得以释放,使得应力应变曲线中出现应力水平突然下降的现象。宫子琪[10]等分析了TiAl基合金断裂韧性的主要影响因素,分析了加载率、温度、环境等因素对TiAl基合金断裂韧性的影响,并没有涉及到Nb对TiAl基合金断裂韧性的影响。
截止目前, 已有的关于γ-TiAl合金单晶的分子动力学研究主要集中在温度、加载速率、裂纹空洞剪切变形、拉伸变形和相变行为等方面,而Nb对裂纹扩展过程及机理的研究尚不多见。用分子动力学方法对裂纹扩展过程进行研究时大多数采用边缘裂纹的单晶γ-TiAl模型,而引入Nb元素模型的未见报道。
针对微观尺度对单晶γ-TiAl合金裂纹扩展研究的不足,本文用分子动力学方法引入加入Nb元素的γ-TiAl模型,预制边界裂纹,通过与单晶γ-TiAl合金原子运动轨迹图与能量演变图相比较的方式,揭示γ-TiAl合金裂纹扩展的机理。
2 模型的建立和计算方法
2.1 模型的建立
由于铌的加入,模型的晶格结构将发生变化,主要表现在改变TiAl合金的晶格常数比c/a,以提高合金的对称性和变形协调性。γ-TiAl合金具有L10型面心四方(fct)晶体结构模型[11],加入铌的γ-TiAl合金仍是四方(fct)结构,但是其c/a增加,即c/a之比由原来的1.03增大至1.045。建立的初始模型如图1所示,通过取消原子间作用力的方法预制边界裂纹,裂纹长度为10a,模拟(010)[100]方向的I型裂纹的扩展过程,模型尺寸为100a×6b×50c,体系共123012个原子。图1(a)中Nb的浓度为3%,为避免原子的热激活效应,温度设置为1K。采用嵌入原子势法(EAM)[12]描述原子间的相互作用力,进行分子动力学模拟,X和Z方向采用自由边界条件,Y方向为周期性边界条件。首先对体系弛豫100ps,达到平衡状态;其次进行拉伸加载,下表面原子固定不动,上表面原子沿Z方向施加3.5×108的恒定应变率。模拟时间每步为0.0001ps,整个程序共运行500万步,每隔2000步记录动能、势能、总能量及应力值。

图1 (a)加入Nb的γ-TiAl合金的原子模型;(b)γ-TiAl 合金的原子模型(图中不同颜色的详细表达请与作者联系)Fig.1 (a) Atom model of γ-TiAl mixed with Nb alloy; (b) Atom model of γ-TiAl alloy
2.2 势函数的选取
常见的势函数有对势(包括L-J势函数、Morse势函数、Jonson势函数等)、多体势(EAM势函数、F-S势函数等)。对势只考虑了原子两两之间的相互作用,并导致了大部分金属晶体并不满足的Cauchy关系,所以其使用具有一定的局限性。相比于对势,多体势考虑了某些重要的多体间相互作用,表述更加精确,故得到了广泛应用。本文在模拟γ-TiAl合金中的裂纹扩展行为时,选择多体势中的EAM嵌入原子势,因为EAM表达式中所用到的具体参数是利用大量可靠的实验数据和第一性原理方法计算拟合得到的,经验证,该势函数可以较好地描述γ-TiAl体系中晶格变形等相关问题[13],因此模拟中可以采用EAM嵌入原子势描述原子间的相互作用力。EAM嵌入原子势将势能分为相互作用对势和嵌入势两部分。相互作用对势就是原子核对它周围原子的原子核的相互排斥力;嵌入势即为原子的核外电子与在电子云背景中的原子的静电作用。此时,系统的总势能为:
(1)
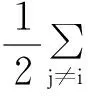
势函数是一种可以表示原子或分子间相互作用的
函数,原子间的相互作用行为从本质上决定着材料的性质,这种相互作用就体现在具体的势函数上。由于研究的是分子尺度的材料性能,所以不能忽视铌的加入对势函数的影响。由于晶格结构的变化导致rij即第i个原子与第j个原子之间的距离增加,将参数进行调整拟合获得合适的势函数。
3 结果与分析
运用LAMMPS软件进行计算,通过OVITO软件进行分析和可视化处理,充分弛豫后裂纹开始扩展,观察原子运动轨迹和能量的演化过程,分析裂纹的扩展机理。
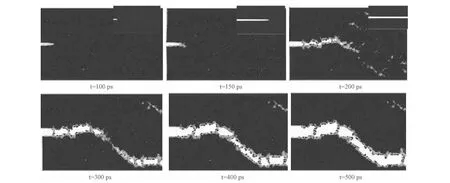
图2 应变率为3.5×108时的裂纹扩展图Fig.2 Crack propagation at 3.5×108 strain rate
图3(a)为未加入Nb元素的γ-TiAl合金裂纹开始扩展及断裂过程中应力随时间的变化曲线,从图3(a)可得出,裂纹开始扩展时的应力值均在5.8GPa左右,应力在裂尖处集中,达到裂纹开始扩展的临界应力值时,裂纹开始扩展,之后应力值随着裂纹的扩展而逐渐衰减,直到断裂时应力值减小为0。图3(b)为加入Nb元素后,γ-TiAl合金裂纹开始扩展及断裂过程中应力随时间的变化曲线,从图3(b)可得出,裂纹开始扩展时的应力值均在6.9GPa左右,与图3(a)相比较,加入Nb元素的γ-TiAl合金应力随裂纹的扩展衰减得非常缓慢,特别是在250ps之后应力随着应变的增加而缓慢减小,材料出现明显的塑性,说明在衰减过程中,应力集中在裂尖前端原子结构出现混乱的地方,随即便萌生了空洞,因此在结构混乱的地方更容易出现应力集中现象。
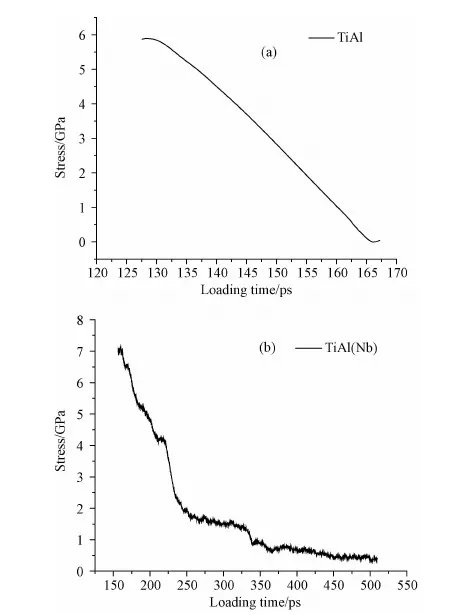
图3 加入Nb后裂纹开始扩展及断裂过程中应力随时间的变化曲线Fig.3 Curve of stress as function of loading time in the process of crack propagation and fracture before and after adding Nb
模拟过程分为弛豫阶段和加载两个阶段。首先将模型在等温等压系综(NPT)下弛豫100ps,以使体系在加载之前达到平衡状态,弛豫过程中三个方向都为周期性边界条件,弛豫之后开始加载,加载时将X和Z方向的边界条件变为自由边界,Y方向仍为周期性边界条件,以减小试件中的残余应力,在加载之前得到符合实际的模拟式样。弛豫阶段总能量随时间的演化过程如图4所示。图4(a)为γ-TiAl合金在弛豫阶段总能量随时间的演化过程图,(b)为加入Nb的γ-TiAl合金在弛豫阶段总能量随时间的演化过程图,总能量达到平衡状态的值分别是-552021ev和-5541095ev,(a)图中弛豫阶段能量先逐渐增加然后趋于平衡,(b)图中弛豫阶段能量先逐渐减小然后趋于平衡,可知Nb的加入对弛豫过程中总能量的变化会产生影响。图5所示为γ-TiAl合金与加入Nb的γ-TiAl合金在加载过程中的总能量随时间的演化过程图。拉伸过程中,初始加载时由于载荷不断增加,内部原子开始运动,动能增加,势能也从平衡态开始上升,γ-TiAl合金内部原子总能量上升,直至出现峰值,此阶段对应γ-TiAl合金的弹性变形阶段。继续施加载荷,模型中的大量原子离开模拟空间,系统的总能量迅速下降,结合原子运动轨迹图可知,此时试件已断裂,图5(a)中γ-TiAl合金能量最终在-550530ev附近趋于平衡,图5(b)中加入Nb的γ-TiAl合金能量最终在-551360ev附近趋于平衡。材料在塑性变形过程中出现的位错、空洞等现象会消耗部分能量,而随着位错的积累又会使能量增加,从而总能量出现明显的上下波动,但是从γ-TiAl合金裂纹扩展的轨迹图中看到裂纹发生完全脆性解理断裂,没有出现位错、空洞现象,这就解释了能量曲线只有一个波峰,且没有出现明显的上下波动现象;然而加入Nb的γ-TiAl合金由于出现位错、空洞现象,因此其能量变化曲线不是只有一个波峰并且出现明显的上下波动现象。
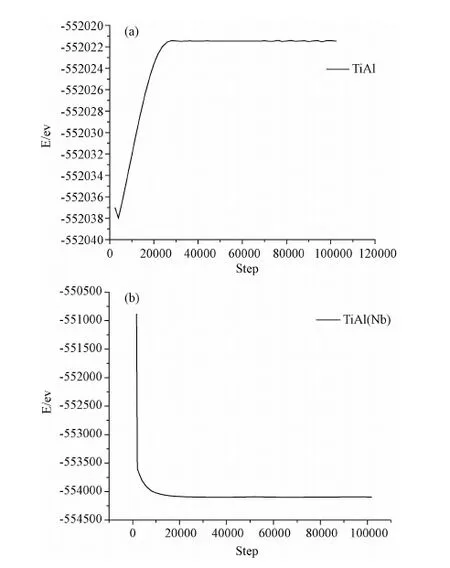
图4 弛豫过程中总能量随时间的演化过程Fig.4 Total energy as function of loading time at relaxation process
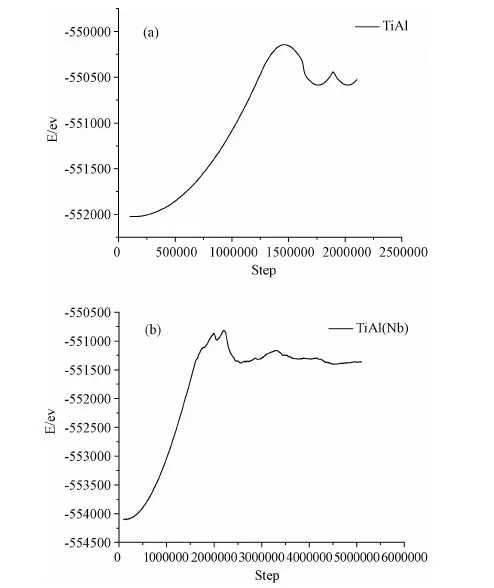
图5 拉伸过程中总能量随时间的演变图Fig.5 Total energy as function of loading time at tensile process

图6 Nb加入前后γ-TiAl合金的应力-应变曲线Fig.6 Stress-strain curves in single crystal γ -TiAl alloy mixed with and without Nb
应力-应变曲线反应材料的力学性能。图6(a)为γ-TiAl合金应力-应变曲线图,(b)为加入Nb的γ-TiAl合金应力-应变曲线图。从图6(a)中可以看出ε=0.042时,试件断裂,只经历了弹性变形阶段,而没有发生塑性变形,波峰应力值5.89GPa。随着应变逐渐增加,原子间作用力逐渐减弱直至消失,应力突然下降。变形初始阶段,应力基本呈线性上升,微观尺度下材料的弹性变形与宏观尺度下的一致。从图6(b)中可以看出,应力-应变曲线变化趋势与(a)图变化趋势基本相同,ε=0.053时,波峰应力值7.13GPa。裂纹扩展之后,应力随应变的增大而逐渐减小,由于应力集中在裂尖前端原子结构出现混乱的地方,随即便萌生了空洞,容易出现应力集中现象,裂纹扩展比较困难,应力减小比较缓慢。
4 结 论
本文用分子动力学方法,研究了预制边界裂纹并加入Nb的单晶γ-TiAl合金的裂纹扩展过程,分析了裂纹在1K温度下的扩展行为,得出以下结论:
1.加入Nb元素后,体系内的原子运动减缓,在相同的应变率条件下,试件的断裂时间延长,说明Nb元素的加入提高了γ-TiAl合金的塑性。
2.加入Nb的γ-TiAl合金,弛豫阶段能量先逐渐减小然后趋于平衡,没有加入Nb的γ-TiAl合金在弛豫阶段能量先逐渐增加然后趋于平衡。
3.Nb元素的加入,出现能量变化曲线有两个波峰并且出现明显的上下波动现象。
4.γ-TiAl合金拉伸过程中应力随着应变均出现先增大后减小的趋势,但是加入Nb元素后,原子之间的内聚力增加,裂纹扩展非常缓慢,随之应力减小缓慢。
[1] Darolia R, et al. Structural Intermetallics[C]. Las Vegas:TMS, 1993, 127.
[2] Kim Y W. Ordered intermetallic alloys partIII: γ-titanium aluminides[M]. JOM, 1994, 46(7):30~39.
[3] Cheol Choia, Hyun Jin Kima, Yong-Tai Leeb, et al. Effects of microstructural parameters on the fatigue crack growth of fully lamellar γ-TiAl alloys[J]. Materials Science and Engineering: A, 2002, 329~331: 545~556.
[4] 付蓉, 芮执元, 等. 单晶 γ-TiAl合金微裂纹扩展行为的分子动力学模拟[J]. 功能材料, 2015, 46(13): 13100~13105.
[5] 蒋孟玲, 李慧中, 刘咏, 等. Nb含量对TiAl 合金铸态组织的影响[J]. 粉末冶金材料科学与工程, 2014, 19(3): 367~372.
[6] 余龙, 宋西平, 张敏, 等. 高铌TiAl合金蠕变变形的原位观察[J]. 稀有金属材料与工程, 2014, 43(4): 881~885.
[7] Xu D S, Wang H, Yang R, et al. Point defect formation by dislocation reaction in TiAl[J]. Materials Science and Engineering, 2009, 3(1): 1~6.
[8] Tang F L, Cai H M, Bao H W, et al. Molecular dynamics simulations of void growth in γ-TiAl single crystal[J]. Computational Materials Science, 2014, 84: 232~237.
[9] 曲洪磊, 王宇, 夏源明. γ-TiAl 单晶纳米杆拉伸变形的分子动力学研究[J]. 中国科学技术大学学报, 2009, 39(6): 627~63.
[10] 宫子琪, 周峰, 柴丽华,等. TiAl合金断裂韧性的影响因素及其韧化机制[J].材料科学与工程学报, 2014, 32(3): 465~468.
[11] Chubb S R, Papaconstantopoulos D A, Klein B M. First-principles study of L10Ti-Al and V-Al alloys[J]. Physical Review B, 1988, 38: 12120.
[12] 张邦维, 胡望宇, 舒小林. 嵌入原子方法理论及其在材料科学中的应用[M]. 长沙:湖南大学出版社, 2003, 75~77.
[13] Zope R R, Mishin Y. Interatomic potentials for atomistic simulations of Ti Al system [J]. Phys RevB, 2003, 68: 024102
[14] Hans W. Horn, William C. Swope. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew [J]. Journal of Chemical Physics, 2004, 120(20):9665~9678.
Effect of Niobium on Crack Propagation in Single Crystal γ -TiAl Alloy Using Molecular Dynamics Simulation
YANG Li1,2, RUI Zhiyuan1,2, LUO Dechun1,2, YAN Changfeng1,2
(1.School of Mechanical and Electrical Engineering College, Lanzhou University of Technology, Lanzhou 730050, China;2.Lanzhou University of Technology digital manufacturing technology and application of Key Laboratory of the China’s Ministry of Education, Lanzhou 730050, China)
With molecular dynamics method the study of the crack propagation in single crystal γ-TiAl if mixed with Niobium in the atomic scale level was carried on. The atomic trajectory figure and energy evolution figure have been obtained by the simulation based on the crack propagation process under the establishment of a crystal structure model of γ-TiAl alloys. The energy and stress-strain relation of the γ-TiAl were analyzed in terms of the effect of niobium on crack propagation. The results indicate that the fracture time is prolonged at the same strain rate, and two crests and significant fluctuations in the energy varying curve can be found after adding niobium element; With the increase of strain, the stress increases firstly then decreases gradually; The growth of crack propagation is slow and the cross section is not smooth with the configuration of crack propagation changing.
Niobium; γ-TiAl alloy; molecular dynamics; crack propagation; energy
1673-2812(2017)03-0503-06
2015-12-14;
2016-02-29
国家自然科学基金资助项目(51065014),甘肃省自然科学基金资助项目(148RJZA008),甘肃省高等学校科研资助项目(2014A-033)
杨 利(1990-),硕士研究生,主要从事材料机械强度的研究。E-mail: Lizzy900912@163.com。
TG146;O77;TG131
A
10.14136/j.cnki.issn 1673-2812.2017.03.031
