Finding chemopreventatives to reduce amyloid beta in yeast
2016-12-02IanG.Macreadie,CostaArvanitis,PrashantBharadwaj
PERSPECTIVE
Finding chemopreventatives to reduce amyloid beta in yeast
Alzheimer’s disease (AD) is the most common form of age-related dementia with the latest report (WorldAlzheimerReport, 2015) showing 46.8 million people are currently affected by dementia. That number is expected to double every 20 years unless there is effective therapeutic intervention.
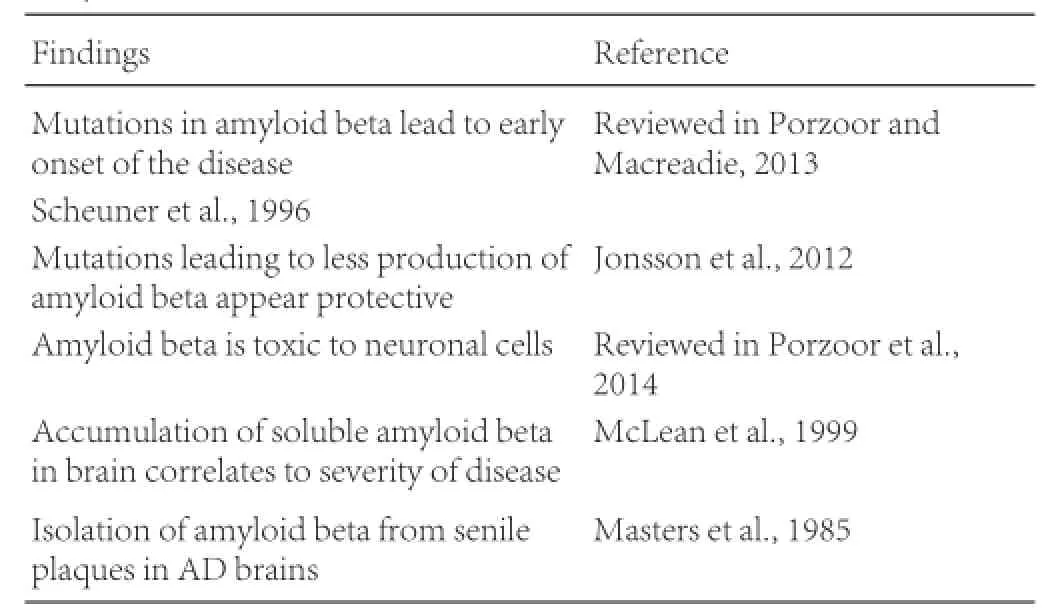
The 42 amino acid peptide known as amyloid beta (or Aβ) has been implicated as one of the main causative agents of AD after its discovery in plaques in 1985 (Masters et al., 1985). Since then evidence has accumulated to support the association between AD and Aβ.
Amyloid beta in AD:
However, progress to find a cure for AD has been very slow, due to lack of reliable models and a lack of understanding about what role Aβ plays in AD (reviewed in Moosavi et al., 2015).
For a period, funding agencies including the Alzheimer’s Association and the Alzheimer’s Drug Foundation were turning away from projects focused on Aβ. To be fair, part of the rationale was probably to diversify their portfolio, but certainly many have questioned the approaches used in Aβ research.
Convincing evidence of Aβ being the cause of AD has been sought through treatments with therapeutic antibodies that bind and remove Aβ. Such treatments include the humanized monoclonal antibodies crenezumab (a homologue of bapineuzamab). Results in clinical trials with bapineuzumab were initially promising and showed clearance of brain amyloid and the lowering of phosphorylated tau protein in the cerebrospinal fluid, however, people did not recover from AD. The failure to cure AD (Panza et al., 2012) led many to doubt that Aβ should be the prime target for the treatment of AD, resulting in a backlash against research on Aβ as a drug target (e.g., see Hyman and Sorger, 2015). The recent antibody trial is showing some promise, although it did not meet the desired end points (Underwood, 2015).
Two non-exclusive hypotheses for the failures are poor bloodbrain barrier penetration (0.1%) and treatment of patients who progressed too far to recover from the disease. Now the tide may turn. A major trial, using people who have no disease but who have biomarkers indicating a likely progression to AD, is addressing the question of whether therapeutic antibodies against Aβ might prevent AD. The early indications are that therapeutic antibodies that clear Aβ can indeed prevent or delay development of AD. Aβ appears like it will be in vogue as a target once again!
Therapeutic antibodies are magic bullets for acute diseases like cancer. They are very expensive but affordable in a wealthy society over the relatively short duration for the treatment of acute diseases. But therapeutic antibodies are not the answer for prevention of a chronic disease like AD. They are simply not affordable, and they appear impractical for extended use. They are important in establishing that Aβ is a valid target for therapy but frequent injections and huge on-going cost means that long term use as a therapy is not viable.
While a description of all the metabolic pathways of Aβ production and clearance is beyond the scope of this perspective, (for a review see Zhang et al., 2011), the well documented beneficial effects of statins can be mentioned. Studies have found up to a 70 % decreased risk of AD in people taking statins and reduced production of Aβ. The stains, originally used as cholesterol inhibitors are now being used to manage neurodegenerative disorders such as vascular dementia and AD. In a research article focusing on effects of Simvastatin in AD, researchers demonstrated a differential dose-dependent effect with Simvastatin on hypoxia inducible factor 1α (HIF-1α) and amyloid precursor protein cleaving enzyme (BACE) in cultured AD cytoplasmic hybrid cells (cybrid cells). These Cybrid cells are being used in research to study dysfunctional mitochondria in AD pathogenesis (Jeong et al., 2015).
There is growing interest in naturally derived compounds as chemopreventatives to remove Aβ. Such compounds should be taken conveniently, which means it is best if they can be ingested. Epidemiology suggests there are many compounds in existing foods that may be useful for chemoprevention of AD (Pandey and Risvi, 2009). Since we have reasonable ideas about the molecular basis of Aβ, it is important that we incorporate these ideas into rational screening for AD chemopreventatives.
What do we know about Aβ and what it does? We know that Aβ accumulates in brains with age. In yeast we have found that young cells, that are newly budded, remove Aβ. Slightly older cells also clear it. But the oldest cells, “grand mothers” retain the Aβ. We know this from in vitro studies where the Aβ is fused to green fluorescent protein (GFP) (see Figure 1). The older cells are readily recognized by the presence of multiple bud scars: one bud scar is left for every cell budded off. In Figure 1, isogenic cells from the same parent can be observed: the older cells with multiple bud scars (stained with calcofluor) have accumulated Aβ and exhibit green fluorescence. It can also be observed that the older cells pass the green fluorescent fusion protein to their progeny, and for a brief time the bud is fluorescent, but it rapidly disappears due to increased autophagy in the young cell.
How do we apply this knowledge? We can screen for compounds that reduce the Aβ related pathology in a cell population. Our detailed methods of doing this are recently published (Porzoor and Macreadie, 2015). Useful compounds (a) increase clearance of Aβ, or (b) reduce Aβ induced toxicity. Such compounds may be useful as chemopreventatives that reduce the level of Aβ, and they may have broader benefits for other age-related protein misfolding diseases. Laterpiridine is a compound that has some history as an AD drug and yeast screening studies show that it increases turnover of Aβ (Bharadwaj et al., 2012). It has also been shown that pure components of green tea, epigallocatechin gallate (EGCG), and salvianolic acid (from the traditional Chinese herb, Danshen) lead to reduced levels of Aβ (Porzoor et al., 2015). Interestingly in vitro studies also show that EGCG and salvianolic acid block fibril formation, suggesting the direct interaction between amyloid beta and these compounds (Porzoor et al., 2015). To explain the in vivo results it would appear that such binding targets the Aβ-salvianolic acid complex, or Aβ-EGCG complex for degradation. An alternate explanation is that salvianolic acid or EGCG might trigger autophagy independently. Either way, the removal of Aβ is the desired outcome.

Figure 1 Amyloid beta persists in older yeast cells.
Aβ is cytotoxic as an oligomeric species, and this characteristic also lends itself to screening. Such screening can be performed using the exogenous addition of chemically-synthesised Aβ. However, recently doubts have been created about the preparation of such peptide (Porzoor et al., 2014). The more common hexafluoroisopropanol method results in toxic peptide, but an ammonium hydroxide preparation results in Aβ that stimulates growth. Which preparation is biologically relevant? Certainly cellular toxicity is widely looked at, but stimulation of growth may be relevant, since neuronal cells are terminally differentiated and do not divide, but in AD many neuronal cells are found to have entered a cell division cells that has led to their death! The more convenient assay is to look for oligomerisation in the Aβ fused to green fluorescent protein (GFP). Monomeric Aβ fused to GFP will exhibit full fluorescence levels, but the dimeric species is not expected to fluoresce. Therefore increasing green fluorescence should also be a good outcome. Indeed this is the basis of assays developed in E. coli, where all fusion protein is non-fluorescent, but fluorescent in the presence of oligomerisation inhibitors (Park et al., 2011).
The ability of yeast to be used as a tool for screening anti amyloidogenic compounds is a useful and unique contribution. It can be adapted to high throughput use, it informs about compounds that are bioavailable, and it requires no Aβ preparation. In a world that seeks new ways to lower Aβ, the yeast assays should be valuable additions to moving forward.
AD chemopreventatives are an economical approach to protect the future aging population. The solution will not be a single compound but is likely to comprise pharmaceuticals and nutraceuticals that not only target Aβ but improve overall cell robustness.
Ian G. Macreadie*, Costa Arvanitis, Prashant Bharadwaj
School of Science and Health Innovations Research Institute, RMIT University, Melbourne, Victoria, Australia (Macreadie IG, Arvanitis C)
Centre of Excellence for Alzheimer’s disease Research and Care, School of Medical Sciences, Edith Cowan University; CHIRI Biosciences Research Precinct, School of Biomedical Sciences, Curtin University, Perth, WA, Australia (Bharadwaj P)
*Correspondence to: Ian G. Macreadie, Ph. D., ian.macreadie@rmit.edu.au.
Accepted: 2015-12-18
orcid: 0000-0001-5335-7220 (Ian G. Macreadie)
Bharadwaj PR, Verdile G, Barr RK, Gupta V, Steele JW, Lachenmayer LM, Yue Z, Ehrlich ME, Petsko G, Ju S, Ringe D, Sankovich SE, Caine JM, Macreadie IG, Gandy S, Martins RN (2012) Latrepirdine (DimebonTM) enhances autophagy and reduces intracellular GFP-Aβ42 levels in yeast. J Alzheimers Dis 32:949-967.
Hyman BT, Sorger P (2015) Failure analysis of clinical trials to test the amyloid cascade hypothesis. Annals Neurol 76:159-161.
Jeong JH, Yum KS, Chang JY, Kim M, Ahn JY, Kim S, Lapchak PA, Han MK (2015) Dose-specific effect of simvastatin on hypoxia-induced HIF-1α and BACE expression in Alzheimer’s disease cybrid cells. BMC Neurol 15:127.
Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Ho K (2012) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488:96-99.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc Natl Acad Sci U S A 82:4245-4259.
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46:860-866.
Moosavi B, Mousavi B, Macreadie IG (2015) Yeast model of Aβ and tau aggregation in Alzheimer’s disease. J Alzheimers Dis 47:9-16.
Pandey KB, Rizvi SI (2009) Plant polyphenols as dietary antioxidants in human health and disease. Oxid Med Cell Longev 2:270-278.
Panza F, Solfrizzi V, Imbimbo BP, Logroscino G (2014) Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: the point of no return? Expert Opin Biol Ther 10:1465-1476.
Park SL, Pegan SD, Mescar AD, Jungbauer LM, Du MJL, Leibman SW (2011) Development and validation of a yeast high-throughput screen for inhibitors of Aβ42 oligomerization. Dis Models Mech 4:822-831.
Porzoor A, Macreadie I (2013) Application of yeast to study the tau and amyloid-β abnormalities of Alzheimer’s disease. J Alzheimers Dis 35:217-225.
Porzoor A, Caine JM, Macreadie IG (2014) Pretreatment of chemically-synthesized Aβ42 affects its biological activity in yeast. Prion 8:404-410.
Porzoor A, Macreadie I (2015) Yeast as a model for aggregation toxicity in Alzheimer’s disease, autophagic responses and drug screening. In: Systems biology of Alzheimer’s disease: Methods and protocols (Castrillo JI, Oliver SG, eds), pp217-226. Humana Press Ch.
Porzoor A, Alford B, Hügel HM, Grando D, Caine J, Macreadie I (2015) Anti-amyloidogenic properties of some phenolic compounds. Biomolecules 5:505-527.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD,Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Vitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, et al. (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Med 2:864-870.
Underwood E (2015) Alzheimer’s amyloid theory gets modest boost. Science 349:464.
WorldAlzheimerReport 2015 Available at http://www.alz.co.uk/research/ WorldAlzheimerReport2015.pdf
Zhang Y, Thompson R, Zhang H, Xu H (2011) APP processing in Alzheimer’s disease. Mol Brain 4:3.
10.4103/1673-5374.177729 http://www.nrronline.org/
How to cite this article: Macreadie IG, Arvanitis C, Bharadwaj P (2016) Finding chemopreventatives to reduce amyloid beta in yeast. Neural Regen Res 11(2):244-245.
杂志排行

中国神经再生研究(英文版)的其它文章
- Tissue-type plasminogen activator is a modulator of the synaptic vesicle cycle
- Impaired consciousness caused by injury of the lower ascending reticular activating system: evaluation by diffusion tensor tractography
- Considering calcium-binding proteins in invertebrates: multi-functional proteins that shape neuronal growth
- Cardiovascular dysfunction following spinal cord injury
- Practical application of the neuroregenerative properties of ketamine: real world treatment experience
- Exergames: neuroplastic hypothesis about cognitive improvement and biological effects on physical function of institutionalized older persons