运动对内质网应激与胰岛素抵抗之间的影响
2016-11-30戈哲
戈 哲
运动对内质网应激与胰岛素抵抗之间的影响
戈 哲
二型糖尿病一直都是威胁人类健康的疾病之一,它的主要特征是胰岛β细胞胰岛素分泌不足和胰岛素抵抗。而内质网应激与胰岛素抵抗之间的联系是最近研究的热点。而大多数研究也表明运动能够缓解二型糖尿病的症状,改善细胞胰岛素抵抗。并且患者经过一系列的耐力运动之后,糖耐量有显著的提高,血糖水平也趋于稳定。众多研究表明,运动可以提高胰岛素受体的敏感性,并且运动与内质网应激也存在一定的关系。本文主要综述近些年来,内质网应激和胰岛素抵抗之间的关系,以及运动在其中扮演着何种角色,从而为相关研究者提供参考借鉴。
运动;内质网应激;UPR;胰岛素抵抗
1 内质网
1.1 内质网应激
内质网是一种膜性细胞器,根据其结构可分为粗面内质网和滑面内质网两种。它内与细胞核外膜相连,外与细胞膜相接,使之成为透过膜连接的一个整体。内质网负责物质从细胞核到细胞质、细胞膜以及细胞外的转运过程。因为细胞内质网膜与细胞核外膜是相连的,因此内质网空腔与核周腔是共通,且细胞可以靠内质网的膜来快速调节细胞核的大小。粗面内质网上附着有大量核糖体,合成膜蛋白和分泌蛋白。光面内质网上无核糖体,为细胞内外糖类和脂类的合成和转运场所。内质网的应激则是内质网周围环境的变化,例如缺氧、化学毒物等多种因素抑制内质网蛋白质加工的糖基化或者导致蛋白质二硫键的错配,甚至内质网的钙离子的大量流失导致未折叠蛋白质的积聚,这些导致内质网功能紊乱的情况,统称为内质网应激。
1.2 线粒体和内质网之间的关系
早在50年前科学家们就观察到两个细胞器有实质上的接触,并且二者之间存在着广泛的信号分子的交流。线粒体和内质网之间存在着精密的连接位点,但是它们并不互相融合,各自维持着各自细胞器独特的结构。随后,随着生化技术的发展,线粒体和内质网连接位点被称作线粒体内质网关联膜(MAMs)。MAMs涉及到钙离子的转运、自噬以及炎性体的形成。内质网钙离子的外流造成了细胞内钙离子的快速增长,由于内质网和线粒体之间的联系,线粒体倾向于摄取高浓度的钙离子,钙离子则从MCU通道进入线粒体,一些位于内质网和线粒体连接位点的蛋白调节钙离子的转运和细胞凋亡,并且还承担着维持线粒体内质网联系结构的功能[1]。
在酵母中,完整的内质网膜蛋白Mmm1p,Mdm12p还有线粒体外膜蛋白Mdm10p和Mdm34p对内质网和线粒体之间的相互关系具有维系的作用[2]。有研究表明线粒体内质网关联膜表面的部分蛋白不仅具有协调钙离子转运和自噬的功能,还具有维持内质网和线粒体正常接触的功能[1]。近来,众多的研究表明MAMs在2个细胞器之间主要承担钙离子的转运过程,这主要是通过MAM上IP3Rs调节来进行完成的,IP3Rs主要调节内质网钙离子的释放[3]。在细胞凋亡的开始阶段,内质网和线粒体之间的钙离子信号变化是凋亡最关键的起始象征。钙离子从内质网向细胞浆和线粒体的转移导致了细胞凋亡[4]。
MAMs之间不仅仅只是存在钙离子的信号交流,脂质分子的交换过程同样也是尤为明显。对于磷脂质,它是最佳的磷脂酰乙醇胺合成的原料之一,磷脂酰丝氨酸在磷脂酰丝氨酸合酶的作用下在内质网合成,然后,磷脂酰丝氨酸被转运到线粒体内膜外表面通过磷脂酰丝氨酸脱羧酶脱羟基生成磷脂酰乙醇胺。最后磷脂酰乙醇胺又返回到内质网,在其被磷脂酰乙醇胺N-甲基转移酶加入甲基基团,从而合成磷脂酰胆碱。磷脂酰丝氨酸合酶专一地定位在MAMs上,它催化过程中的限速步骤就是磷脂酰丝氨酸向线粒体的转移。并且,MAMs上有多种胆固醇和神经酰胺合成所必需的酶类[5]。
并且,线粒体和内质网之间联系与自噬体的形成也存在着一定的关系。例如:在饥饿状态下,自噬前体标志物ATG14和DFCP1以及ATG5转移到MAMs上,参与自噬体的形成。并且通过抑制ATG14和DFCP1在MAMs在其位点蛋白的共同定位,例如MFN2和PACS2,进而阻止了正常自噬体的形成,这表明MAMs与自噬体的形成也存在着密切的关系,也充分说明了内质网和线粒体之间存在着多方面的信息交流[6]。
2 UPR
2.1 UPR简介
1988年,Kozutsumi等就提出了内质网应激存在信号转导的过程[7]。未折叠蛋白反应(UPR)是体内出现错误折叠蛋白进而刺激一些列信号通路产生相应的一些列反应来保持细胞内环境稳态的一个过程。它的信号主要是由内质网膜上三种跨膜蛋白所介导,主要是双链RNA 依赖蛋白激酶样内质网激酶(PERK)、肌醇需求激酶(IRE-1α)、活化转录因子(ATF6)。这3个信号分子是通过与分子伴侣Bip之间相互作用而引发的。
2.2 UPR主要的信号通路
2.2.1 PERK 蛋白激酶R样内质网激酶(PERK)是位于内质网膜上的一种I型跨膜蛋白,属于elF2α上游激酶家族中的一种。PERK信号通路的激活发生在内质网应激早期,通过抑制蛋白质的合成对细胞起保护作用、促进细胞的生存。随着内质网应激时间的延长,PERK通过诱导CHOP的表达而促进细胞凋亡。PERK特异性的磷酸酶去磷酸化抑制PERK,调控的信号途径在中枢神经系统、代谢异常等病理过程中发挥着重要的作用。
2.2.2 IRE1α 肌醇依赖性激酶(IRE1α)是一种Ⅰ型跨膜蛋白激酶/RNA 酶,定位于内质网。IRE1有2种分别为IRE1α 和 IRE1β。这两种蛋白互为同系物。IRE1α广泛存在,尤其是在胰腺中,而IRE1β只存在于肠上皮细胞。无论是IRE1α还是IRE1β的过表达都会上调 ER的分子伴侣。在感受到内质网应激信号后,发生二聚化,激活其RNA 酶活性,进而剪接、激活下游分子X-盒结合蛋白1(XBP1)。XBP1作为转录因子继续激活其下游基因从而缓解内质网应激。在胰岛细胞中,IRE1α还可以不依赖于XBP1 而直接降解胰岛素mRNA。
2.2.3 ATF6 活化转录因子6(ATF6)为内质网上的一种感受蛋白,是内质网应激引起的细胞凋亡和自噬途径中的一个重要的调节因子。
2.3 UPR信号通路与自噬
在真核细胞中,由于ER上3种跨膜蛋白PERK、IRE-1αATF6与分子伴侣Bip是处于结合状态的,3种跨膜蛋白在正常情况下是处于无活性的状态的。一旦未折叠蛋白在细胞中堆积过多,Bip则与未折叠蛋白结合从而导致它们被激活。Bip在正常组织中表达水平较低。当错误折叠蛋白在内质网腔内蓄积,与BiP 结合增多,使感应蛋白PERK、IRE1 和ATF6 与BiP解离而被激活,启动UPR,促使UPR介质被释放。
导致UPR的3种主要信号通路:PERK引发elF2的α亚基Ser51发生磷酸化,从而使核糖体的蛋白质组装功能被抑制,通过阻断mRNA的翻译来减缓蛋白质折叠的压力。ATF6则在内质网应激的情况下从Bip结合中释放出来,进而进入高尔基体中,然后被S1P和S2P酶水解,并且其中的N端的ATF6作为转录因子和ERSE结合,从而介导更多ER分子伴侣的产生,增强内质网蛋白质的折叠能力。在 ERS 时,一旦被激活,IRE1α的核酸内切酶活性就会剪去XBP1 mRNA的26 bp内含子,生成XBP1 mRNA 编码蛋白XBP1s,它能调控内质网蛋白质的成熟以及转运,也能调控转录水平上未折叠的蛋白质的降解。具体UPR信号通路如图1所示。
这3种信号通路不仅对蛋白质翻译以及相应mRNA翻译和提高内质网折叠能力有影响。而且还可以通过内质网相关的降解途径(ERAD)和自噬二种方式来降解体内未折叠蛋白或错误折叠蛋白。IRE1α-XBP1通路能活化ERAD的相关降解蛋白,ERAD通路通常用于清除积聚在内质网的小部分可溶性未折叠蛋白,对于大型未折叠蛋白的积聚和损伤的细胞器的降解,ERAD通路则是无能为力,需要依靠自噬的过程来完成。自噬是将体内不需要的蛋白质转运到溶酶体降解进而循环的一个过程,UPR可以激活自噬。PERK-elF2α通路是内质网应激导致自噬最为关键的一个过程,而elF2α磷酸化的增加会导致ATF4翻译的增加,从而导致ATF4表达增多。ATF4和CHOP调节着似乎12个以上的ATG基因,从而引发自噬过程[8]。ATF4还能激活CHOP从而引发细胞凋亡程序,这取决于内质网应激的程度[9]。并且,IRE1似乎也与自噬的激活有关系。TRAF2依赖性激活的IRE1-JNK通路导致了Bcl-2磷酸化,进而导致自噬体的激活[10]。
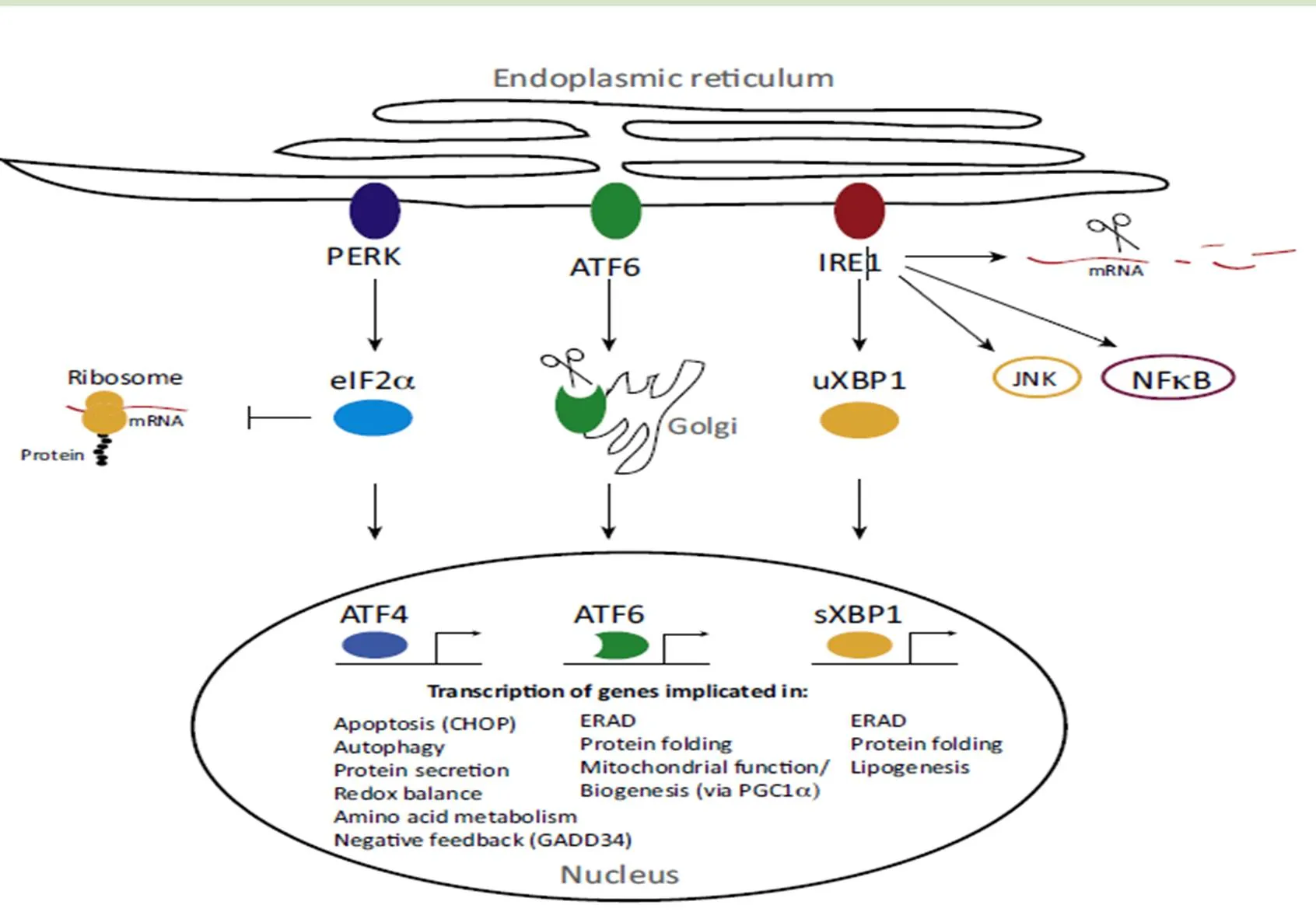
图1 未折叠蛋白反应信号通路
UPR的发生是与细胞内环境的变化有着重大关联的,比如自噬、细胞缺氧、线粒体生物发生和ROS等。饥饿状态、mTOR信号通路、未折叠蛋白的积累都能够导致自噬的发生[11]。细胞缺氧和ROS的产生导致内质网所处的胞浆外环境产生变化,从而导致内质网应激,进而引发蛋白质折叠错误,导致了UPR的产生。线粒体生物发生主要由PGC-1α诱导,而有研究说明PGC-1α与ATF6之间存在着某种联系[12]。多数文献表明,激鉴于这些细胞内环境发生的变化,UPR与人类疾病相关联,例如癌症、糖尿病、神经退行性疾病等。
3 内质网应激与二型糖尿病之间的关系
3.1 二型糖尿病与内质网和线粒体之间的关系
既然蛋白质的折叠如此重要,那么不难推断蛋白质的错误折叠可能会引发某些疾病的发生。我们已经知道,内质网和线粒体之间不论是功能上还是结构上都存在着紧密的联系,但是似乎二者之间与二型糖尿病的关系也同样密切。二型糖尿病是胰岛素抵抗和胰岛β细胞分泌不足的一种代谢紊乱疾病。有趣的是,内质网应激所导致的肝细胞胰岛素抵抗和胰岛β细胞线粒体功能的改变暗示着自噬功能的缺陷。因此,自噬功能的紊乱可能涉及细胞器的功能下降,从而导致二型糖尿病的发生[13],并且线粒体功能的异常导致胞浆游离钙离子浓度的提升进而导致内质网应激,异常的胰岛素信号和肝细胞糖异生的增加[14]。MAMs中钙离子的转换在胰岛素抵抗之中似乎起着至关重要的作用,有研究表明,在糖尿病大鼠肝细胞中,MAMs完整性的损坏将可能通过损伤线粒体或者是诱发内质网应激来引发胰岛素抵抗。而且,线粒体和内质网二个细胞器之间的功能改变将会诱发肝细胞脂质的堆积,例如甘油二酯、神经酰胺、脂酰辅酶A等。这些脂质堆积将会导致丝氨酸/苏氨酸激酶的激活,例如JNK、PKC等,从而进一步导致胰岛素信号通路的抑制。并且,MAMs完整性的损坏将会影响内质网钙离子向线粒体转移,干扰细胞质钙离子浓度,这将会影响钙离子敏感性激酶和胰岛素信号通路相关的磷酸酶的活性,例如CaMKII和钙调磷酸酶[15]。损伤的钙离子稳态将会引发内质网应激和抑制线粒体氧化代谢,因此这将直接导致胰岛素信号通路的抑制或者间接通过脂质积累的过程来抑制。而且,由于MAMs存在着许多的氧化应激蛋白,所以MAMs上面的氧化应激反应对胰岛素的信号调节起着不容忽视的作用[16]。所以,由此我们可以知道,线粒体的损伤、氧化应激、内质网应激、脂质堆积和钙离子稳态彼此都相互之间影响着,并且它们也通过直接或者间接的方式共同调节胰岛素信号通路。
3.2 内质网应激与胰岛素抵抗
胰岛素的信号通路是一个酪氨酸磷酸化的级联反应:首先是胰岛素受体酪氨酸激酶的自身活化,其后是胰岛素受体底物-1 (IRS-1)的酪氨酸磷酸化作用,进而再引发一系列信号分子的级联反应,导致葡萄糖被摄入细胞内加以利用,从而达到降低血糖的效果。体外实验表明,ER 应激可引起包括肝脏、肌肉和脂肪等外周组织的胰岛素抵抗[17]。
代谢和内分泌功能对脂肪组织胰岛素抵抗有一定的影响作用。脂肪组织产生瘦素和脂联素调节食物摄入和分泌抗炎脂肪因子进行调节炎症反应和胰岛素抵抗。炎症反应在脂肪组织中增加了促炎性细胞因子,包括TNF-α、IL-6、1L-1b、CC趋化因子配体2。这些炎症因子造成氧化反应和内质网应激,进而导致胰岛素抵抗[18]。最近有研究表明内质网应激也降低脂蛋白的表达,脂滴包被蛋白A也提高脂肪细胞的分解代谢,内质网应激也激活脂肪分解反应,并且这个过程还能被JNK,p38MAPK and PKC这3种蛋白所抑制,这表明内质网应激能引发脂肪在细胞的堆积,说明了这3种蛋白并且与胰岛素信号调节存在着一定的关系[19]。最近还有数据表明,高同型半胱氨酸(HHcy)能够导致脂肪组织的胰岛素抵抗,HHcy通过蛋白激酶B(PKB)激活JNK从而导致胰岛素抵抗[20]。
Ozcan等[21]利用衣霉素诱导肝细胞ER应激,发现胰岛素刺激的Akt磷酸化和IRS-1酪氨酸磷酸化受到抑制,同时IRS-1丝氨酸磷酸化增强。与酪氨酸磷酸化相比,胰岛素受体或其下游信号配体的丝氨酸磷酸化阻止了胰岛素信号的传导,从而降低了胰岛素在外周组织的敏感性,导致胰岛素抵抗。SREBP-1C(固醇调节元件结合蛋白)它是脂质基因转录激活因子,它被内质网应激所激活。在持续的内质网应激的过程中,SREBP-1c通过暴露在高尔基体上的信号位点释放GRP-78,GRP-78然后转移到高尔基体上,在高尔基体上,SREBP-1c产生活性片段,进而进入细胞核刺激脂肪生成相关基因[22]。有研究表明,在肥胖大鼠肝细胞内过表达GRP78减缓内质网应激降低了SREBP-1c的激活、肝脏甘油三酯在肝脏中的堆积,从而提升了胰岛素的敏感性[23]。TRB3是一种内质网应激诱导蛋白,它是通过内质网应激通路中PERK-ATF4通路所诱发的,它能抑制胰岛素信号通路[24],更有研究表明它的表达在糖尿病大鼠肝细胞中被上调[25][26]。肝细胞TRB3的敲除显著提升糖耐量,它的过表达将导致胰岛素抵抗[27]。
然而,最近Shulman’s团队研究表明IRE-1所导致的JNK的激活与肝细胞胰岛素抵抗之间联系并不紧密。他们展现了在高胰岛素血症下,虽然内质网应激相关分子和JNK的激活,但一周果糖喂养的XBP-1敲除的小鼠仍然展现出升高的肝细胞胰岛素敏感性[28]。并且,脂肪和脂多糖激活的PKR,然后直接通过IRS-1丝氨酸位点或者JNK抑制胰岛素信号通路[29][30],这似乎又表明内质网应激与胰岛素抵抗之间的关系不是像一对一关系那样的明确,具体机制仍然有待研究。
并且在肝细胞内质网应激中,脂肪的堆积并不起主要作用。研究表明,CHOP基因敲除的小鼠,虽然它们仍然是肥胖,但仍然表现出正常的糖耐量和胰岛素敏感性[31]。在肌肉细胞中,高水平的游离脂肪酸将会导致胰岛素抵抗。体内脂质的产生可以调节胰岛素响应和导致糖代谢的缺失和胰岛素的抵抗,例如甘油二酯、脂酰辅酶A、神经酰胺等[32]。近来还有研究表明人类和啮齿动物骨骼肌细胞细胞的胰岛素抵抗主要是由棕榈酸盐转运到细胞内,生成神经酰胺,而神经酰胺通过PKCζ/PP2A的激活来抑制PKB磷酸化来抑制胰岛素信号通路。这也表明了,肌细胞的内质网应激过程似乎与其胰岛素抵抗的关系不大[33]。所以综上所述,在肝细胞中,内质网应激引发的UPR信号通路似乎与胰岛素抵抗存在着密切的联系,而对于肌肉细胞而言,棕榈酸盐在肌肉细胞里的堆积所引发的胰岛素抵抗则似乎起着主要的作用。
4 运动与内质网应激
4.1 运动对内质网应激的影响
运动针对肥胖和二型糖尿病已经展现出非常良好的治疗效果,然后其具体的分子机制仍然是不太清楚的。内质网蛋白质的折叠过程可能被很多其它因素所影响,比如内质网钙离子的耗竭、能量营养的消耗。有研究揭示了运动所引发的PGC1-α与ATF6α之间的联系是调节UPR的主要因素之一,PGC1-α和ATF6α之间存在着直接的接触更加证明了这个观点。有研究表明PGC1-α能够显著上调肌细胞中UPR相关基因的表达,并且在ATF6-α-/-的肌肉细胞中,PGC1-α所导致的UPR信号基因表达的增加出现缺陷,但其具体的机制尚不明确。PGC1-α表现出调控ATF6-α在某一种特定的方式。ATF-6α的辅激活作用可能并不是PGC1-α和UPR之间唯一的通路。值得注意地是,有研究表明,另一种转录共激活因子CRTC2,它通过与ATF-6α或者CREB参与调节肝细胞UPR与糖异生之间的信号通路[34]。这也说明了PGC-1α与UPR之间存在着多条信号通路。一次性运动刺激能够上调UPR反应,而长期的耐力运动则会降低UPR反应,使UPR相关的信号分子有所下调,并且最近有研究表明,UPR会对长期的运动的刺激产生适应效果,这种适应是作为损伤控制机制来保护机体的[35]。而且,有研究表明长期耐力训练将会通过降低PERK和elF2α的磷酸化降低内质网应激程度[36]。所以,我们很容易就可以想到,运动是否也是通过降低内质网应激从而达到缓解胰岛素抵抗的治疗效果呢?
4.2 运动与内质网应激和胰岛素抵抗之间的关系
前面提到衣霉素所诱导肝细胞ER应激,发现胰岛素刺激的Akt磷酸化和IRS-1酪氨酸磷酸化受到抑制,同时IRS-1丝氨酸磷酸化增强,从而导致了胰岛素抵抗。并且在给予JNK 抑制剂SP600125或JNK抑制肽则显著导致了胰岛素抵抗和葡萄糖耐量均有所改善。并且内质网应激的状态下,IRE1α敲除的成纤维细胞的作用下,JNK的活性并未得到增强,并且胰岛素所导致的IRS-1酪氨酸磷酸化增强,这也说明了胰岛素受体敏感性得到了改善[21]。由此我们便可以推测,在肝细胞中,内质网应激可能是通过IRE1α-JNK通路来抑制Akt的磷酸化和促进IRS-1丝氨酸磷酸化,进而起到影响胰岛素信号转导的作用,导致IR。那么运动到底与内质网应激和胰岛素抵抗存在着什么样的关系呢?通过文献查阅直接描述关于运动对二者之间的关系的研究目前还非常少。首先,Williamson研究发现抗阻运动将会降低中老年男性细胞中JNK的磷酸化,从而降低胰岛素抵抗[37]。由此可见运动似乎是通过降低JNK蛋白上的位点磷酸化,从而来降低胰岛素抵抗的。而我们清楚,JNK是IRE1α的下游信号分子,而IRE1α是内质网应激的首先引发分子,由此我们可以推想,运动是否是通过降低内质网应激程度,降低JNK的上游IRE1α的磷酸化来达到降低胰岛素抵抗的目的。而且上面也提到,长期耐力运动似乎能够降低PERK和elF2α的磷酸化水平,而众多文献表明PERK的磷酸化水平是判断内质网是否应激的敏感信号分子。所以我们可以推测运动应该是通过降低内质网应激从而降低胰岛素抵抗。并且还有研究表明,游泳耐力运动能够减缓内质网应激与胰岛素抵抗,具体表现在游泳训练施加在膳食诱导的肥胖大鼠中,大鼠脂肪细胞和肝细胞内质网应激相关分子PERK、eIF2α磷酸化的降低,从而胰岛素受体敏感性增加[36]。
5 小 结
综上所述,线粒体和内质网之间存在着密切的物质交流,尤其是钙离子的平衡。钙离子的紊乱将直接导致内质网合成加工蛋白质的环境发生改变,从而诱发胰岛素抵抗。并且内质网应激也可以通过炎症反应来诱发,从而导致胰岛素抵抗。这也表明了导致内质网应激的因素是多方面的。此外,脂质和蛋白质的中间代谢产物也能直接导致胰岛素抵抗,并不通过内质网应激信号通路。这也说明导致胰岛素抵抗的因素有很多种,内质网应激或许只是其中一个关键的因素。从内质网应激所引发的信号通路来看,内质网应激是通过IRE-1α的激活导致JNK的募集从而导致IRS1丝氨酸位点的磷酸化,进而导致肝细胞胰岛素信号通路的抑制。而且,PERK-elF2α通路似乎通过生成TRB3来抑制PKB靶蛋白来印制胰岛素信号通路。而且,肌肉细胞中的内质网应激似乎和内质网应激关系不大,棕榈酸盐在肌细胞内的直接堆积似乎起着主要的作用。而肝细胞中,内质网应激信号通路的激活所导致的胰岛素抵抗则起着主要的作用。结合运动来看,部分文献表明慢性运动似乎通过降低内质网应激从而达到缓解胰岛素抵抗的作用,但是具体运动是通过何种机制来降低内质网应激程度,目前仍不清楚,有待进一步研究说明。
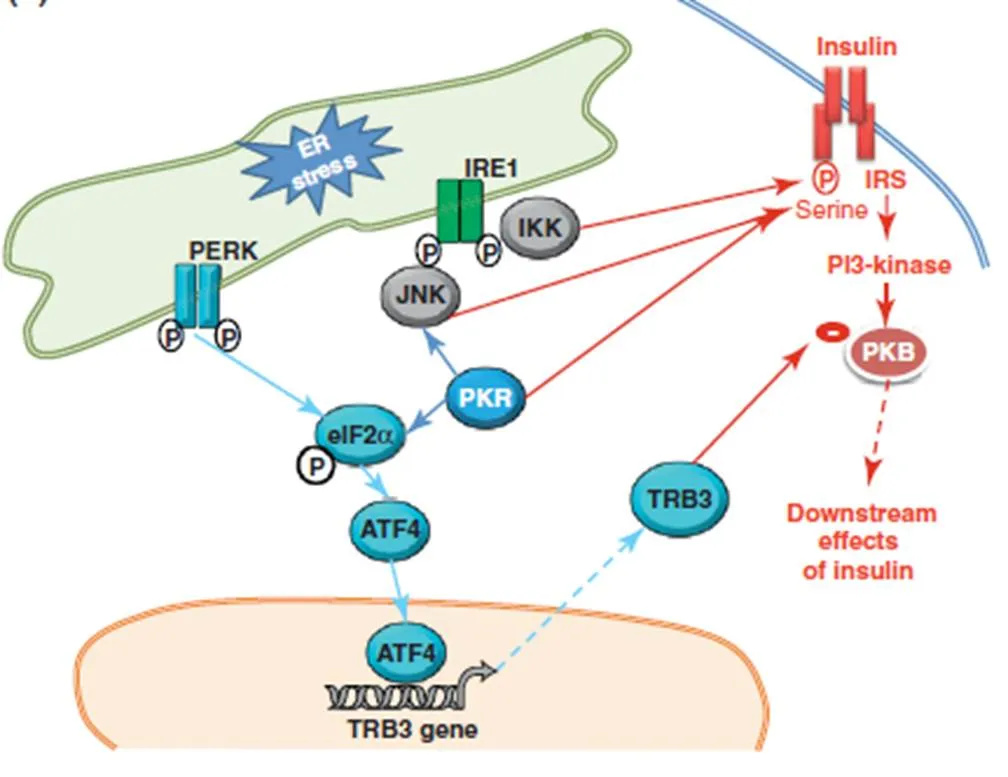
图2 内质网应激对胰岛素信号通路影响机制
[1] Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum–mitochondria connection: One touch, multiple functions[J]. Biochimica Et Biophysica Acta. 2013, 1837(4): 461~469.
[2] Kornmann B, Currie E, Collins S R, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen[J]. Science. 2009, 325(5939): 477~481.
[3] Mikoshiba K. The IP3 receptor/Ca2+ channel and its cellular function[J]. Biochem Soc Symp. 2007(74): 9~22.
[4] Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria[J]. Biochem J. 1987, 245(3): 915~918.
[5] Bionda C, Portoukalian J, Schmitt D, et al. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria- associated membrane) and/or mitochondria?[J]. Biochem J. 2004, 382(Pt 2): 527~533.
[6] Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites[J]. Nature. 2013, 495(7441): 389~393.
[7] Kozutsumi Y, Segal M, Normington K, et al. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins[J]. Nature. 1988, 332(6163): 462~464.
[8] B'Chir W, Maurin A C, Carraro V, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression[J]. Nucleic Acids Res. 2013, 41(16): 7683~7699.
[9] Sano R, Reed J C. ER stress-induced cell death mechanisms.[J]. Biochimica Et Biophysica Acta. 2013, 1833(12): 3460~3470.
[10] Deegan S, Saveljeva S, Gorman A M, et al. Stress-induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress[J]. Cellular & Molecular Life Sciences Cmls. 2012, 70(14): 2 425~2 441.
[11] Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues[J]. Cell. 2011, 147(4): 728~741.
[12] Wu J, Ruas J L, Estall J L, et al. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC- 1alpha/ATF6alpha complex[J]. Cell Metab. 2011, 13(2): 160~169.
[13] Hur K Y, Jung H S, Lee M S. Role of autophagy in beta-cell function and mass[J]. Diabetes Obes Metab. 2010, 12 Suppl 2: 20~26.
[14] Lim J H, Lee H J, Ho J M, et al. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance[J]. Cell Signal. 2009, 21(1): 169~177.
[15] Lale O, Jane C D S, Alp Avi H, et al. Activation of Calcium/ Calmodulin-Dependent Protein Kinase II in Obesity Mediates Suppression of Hepatic Insulin Signaling.[J]. Cell Metabolism. 2013, 18(6): 803~815.
[16] Tiziana A, Leda B, Eva M, et al. Ero1α regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface(MAM).[J]. Antioxidants & Redox Signaling. 2012, 16: 1 077~1 087.
[17] 余万桂,汪长华.内质网应激与胰岛素抵抗的研究进展[J]. 长江大学学报(自科版)医学卷. 2009(03).
[18] Xu L, Spinas G A, Niessen M. ER stress in adipocytes inhibits insulin signaling, represses lipolysis, and alters the secretion of adipokines without inhibiting glucose transport[J]. Horm Metab Res. 2010, 42(9): 643~651.
[19] Deng J, Liu S, Zou L, et al. Lipolysis response to endoplasmic reticulum stress in adipose cells[J]. J Biol Chem. 2012, 287(9): 6 240~6 249.
[20] Li Y, Zhang H, Jiang C, et al. Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue[J]. J Biol Chem. 2013, 288 (14): 9 583~9 592.
[21] Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes[J]. Science. 2004, 306 (5695): 457~461.
[22] Wolins N E, Brasaemle D L, Bickel P E. A proposed model of fat packaging by exchangeable lipid droplet proteins[J]. FEBS Lett. 2006, 580(23): 5 484~5 491.
[23] Kammoun H L, Chabanon H, Hainault I, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest 119[J]. Journal of Clinical Investigation. 2009, 119(5): 1 201~1 215.
[24] Hayashi N O S Y. TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death[J]. EMBO. 2005, 24: 1 243~1 255.
[25] Du K, Kulkarni R N, Montminy M, et al. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver.[J]. Science. 2003, 300 (5625): 1 574~1 577.
[26] Oberkofler H, Pfeifenberger A, Soyal S, et al. Aberrant hepatic TRIB3 gene expression in insulin-resistant obese humans.[J]. Diabetologia. 2010, 53(9): 1 971~1 975.
[27] Koo S H, Satoh H, Herzig S, et al. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3.[J]. Nature Medicine. 2004, 10(5): 530~534.
[28] Jurczak M J, Lee A H, Jornayvaz F R, et al. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice.[J]. Journal of Biological Chemistry. 2012, 287(4): 2 558~2 567.
[29] Urano F, Wang X, Bertolotti A, et al. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1[J]. Science. 2000, 287(5453): 664~666.
[30] Nakamura T, Furuhashi M, Li P, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis.[J]. Cell. 2010, 140(3): 338~348.
[31] Maris M, Overbergh L, Gysemans C, et al. Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance[J]. Diabetologia. 2012, 55(4): 1 167~1 178.
[32] Kraegen E W, Cooney G J. Free fatty acids and skeletal muscle insulin resistance[J]. Curr Opin Lipidol. 2008, 19(3): 235~241.
[33] Hage H R, Hainault I, Vilquin J T, et al. Endoplasmic reticulum stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells[J]. Diabetologia. 2012, 55(1): 204~214.
[34] Wenz T, Rossi S G, Rotundo R L, et al. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging[J]. Proc Natl Acad Sci U S A. 2009, 106(48): 20 405~20 410.
[35] Rutkowski D T, Hegde R S. Regulation of basal cellular physiology by the homeostatic unfolded protein response[J]. J Cell Biol. 2010, 189(5): 783~794.
[36] Da L G, Frederico M J, Da S S, et al. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats[J]. Eur J Appl Physiol. 2011, 111(9):
2 015~2 023.
[37] Williamson D, P G, M H, et al. Mitogen-activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle[J]. Journal of Physiology. 2003, 547(3): 977~987.
Influence of Exercise Between the Endoplasmic Reticulum Stress and Insulin Resistance
GE Zhe
Type 2 diabetes has been one of the disease threat to human health whose main characteristic is islet beta cells insulin hypo secretion and insulin resistance. The connection between the endoplasmic reticulum stress and insulin resistance is a hotspot of recent research. Furthermore, most of the research has shown that exercise can relieve the symptoms of type 2 diabetes, improving the condition of cells insulin resistance. After a series of endurance sports, patients have significant improvement of glucose tolerance and stabilization of blood sugar levels. Numerous studies have shown that exercise can improve the sensitivity of insulin receptor and exercise also has certain relations with endoplasmic reticulum stress. This article mainly reviews the relationship between the endoplasmic reticulum stress and insulin resistance and the role the exercise play in it in the most recent years, providing a reference for related researchers.
Exercise; Endoplasmic reticulum stress; UPR; Insulin resistance
1007―6891(2016)02―0029―06
10.13932/j.cnki.sctykx.2016.02.09
G804.22
A
2015-11-15
华东师范大学体育与健康学院,上海,200241。
Institute of P.E. and Health, East China Normal University, Shanghai, 200241, China.
