X1∑+ 态NaX(X=Br,F)分子势能函数与光谱常数
2016-08-08赵淑令沈光先
赵淑令,吕 兵,沈光先
(贵州师范大学 物理与电子科学学院,贵州 贵阳 550001)
X1∑+态NaX(X=Br,F)分子势能函数与光谱常数
赵淑令,吕兵,沈光先*
(贵州师范大学 物理与电子科学学院,贵州 贵阳550001)
摘要:采用群论及分子反应静力学原理,推导其NaBr和NaF分子基态电子态和合理的离解极限。运用Gaussian03程序中的多种方法和基组,计算分子的几何结构、离解能以及谐振频率,对NaBr分子选取B3LYP/6-311g(2df,2pd)为最优方法与基组,对NaF分子选取BP86/6-31g(3df,3pd)为最优方法与基组,分子在最优方法与基组下进行能量扫描,采用Murrell-Sorbie函数进行了非线性最小二乘法拟合得到分子势能函数,从而计算NaBr和NaF分子的力常数与光谱常数,与实验值对照,计算得到光谱常数值与实验值吻合甚好。
关键词:NaBr;NaF;势能函数;光谱常数;Murrell-Sorbie
0引言
分子势能函数是在Born-Oppenhimer近似下包括电子运动和核运动分子总能量的本征函数,也是在全空间范围内对分子性质的完全描述,即描述分子的能量、几何性质、力常数以及光谱常数,同时,分子势能函数也是研究原子与分子碰撞与反应动力学的基础[1]。近年来,随着天体物理学、分子生物学、激光物理、光化学以及纳米技术的不断发展,分子势能函数尤其是双原子分子的势能函数的研究已成为一个十分重要的研究课题,对双原子分子光谱的研究也成为一个十分重要的研究方向[2-4]。
碱金属的卤化物在工业生产中具有非常广阔的运用前景,NaBr由于具有较好的感光性能,主要用于摄影技术中作为胶片的感光液,最近几年,NaBr不仅仅是运用于摄影技术,而在发光探测、医学以及化学工业方面广泛运用,施毅敏等人利用基于密度泛函理论(DFT)的广义梯度近似(GGA)方法通过计算NaBr分子的介电函数、反射率、能量损失函数、光吸收系数、光导率、折射率等相关数据,研究了NaBr分子的电子结构以及光学性质[5]。崔玉亭等人利用扩展休克尔方法计算了NaBr分子的能带结构[6]。NaF由于具有宽的光学带隙和色心等优良性质在农药制备、金属冶炼工业以及生物医药工业等领域备受关注,在有关核能实验中NaF薄膜材料由于具有防辐射功能而扮演了十分重要的角色。除此之外,NaF还可以作为光电材料以及光电材料的辅助材料,因此研究NaF分子相关的光学以及动力学性质具有十分重要的意义。詹勇军等人从NaF薄膜的制备入手,通过运用原子力显微镜(AFM)、X射线衍射(XRD)和X射线光电子能谱(XPS)分析了NaF薄膜的成分以及表面形貌以及薄膜的微观结构和应力情况[7]。但对其分子的势能函数与光谱数据相关研究报道甚少,研究主要通过从原子与分子的静力学理论[8]出发,推导分子可能电子状态以及离解极限,运用Gaussian03软件计算了NaBr和NaF分子的平衡核间距、离解能以及谐振频率,同时进行单点能扫描,将扫描结果运用origin8.5软件采用Murrell-Sorbie势函数(简称:M-S势)进行拟合,得到分子的势能函数,从而计算分子的力常数、光谱常数,进一步将计算结果与实验值进行对照。
1理论计算
1.1基态NaBr分子的离解极限


1.2分子结构与势能函数
运用Gaussian03软件包,运用密度泛函理论(Density Functional Theory, DFT)中B3LYP、BP86方法以及单双取代耦合族理论(CCSD)和二次组态相互作用方法(QCISD),同时配合6-31g(3df,3pd)、6-311g(2df,2dp)以及6-311++g(3df,3pd)基组计算NaBr和NaF分子的几何结构、离解能以及谐振频率,所计算的平衡核间距、离解能、谐振频率列于表1中。与实验值进行对照,对NaBr分子选取B3LYP/6-311g(2df,2pd)为最优方法与基组,对NaF分子选取BP86/6-31g(3df,3pd)为最优方法与基组。
对NaBr和NaF分子在选取的最优方法与基组下进行单点能扫描。NaBr分子在0.12~0.58nm范围内进行能量扫描。NaF分子在0.06~0.61nm范围内进行能量扫描。所有分子在扫描过程中除核间距在不断变化之外,其它参量均与优化时候参量严格保持一致,运用Origin8.5软件拟合M-S势函数。即:

表1 NaBr分子的平衡核间距、离解能
V=-De(1+a1ρ+a2ρ2+a3ρ3)exp(-a1ρ)
(1)
式中ρ=R-Re,R和Re是双原子分子的核间距和平衡核间距,相应地,De是离解能,a1,a2,a3是双原子分子的势能函数拟合参数,拟合参数列于表2中,拟合势能曲线如图1所示。分子的离解能De由分子的零点能经(2)式修正得到,即
(2)

图1 基态NaBr分子M-S势能曲线Fig.1 The M-S potential energy curve of curve the ground states NaBr

图2 基态NaBr分子M-S势能曲线Fig.2 The M-S potential energy of the ground states NaF

M-Sa1/nm-1a2/nm-2a3/nm-3f2(aJnm-2)f3(aJnm-3)f4(aJnm-4)NaBr14.517227.0683350.802993.59-3505.4111755NaF19.274477.2086821.6354181.16-8613.9375468
1.3NaBr分子力常数与光谱常数
NaBr分子力常数与光谱常数采用(3)~(9)式进行计算,所计算得到的力常数列于表2中,NaBr和NaF分子的光谱常数分别列于表3、表4中。
(3)
(4)
(5)
(6)
(7)
(8)
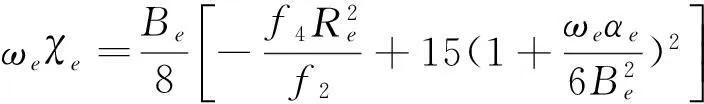
(9)
其中,f1、f2、f3分别为二阶、三阶、四阶力常数,μ为折合质量,c为光速,ωe和ωeXe分别为谐振频率和非谐振频率,Be和αe分别为刚性转动因子和非刚性转动因子。

表3 NaBr分子的光谱常数

表4 NaF分子的光谱常数
2结论
采用Gaussian03程序中密度泛函理论中B3LYP、BP86方法和单双取代耦合族理论(CCSD)和二次组态相互作用方法(QCISD),同时配合配合6-31g(3df,3pd)、6-311g(2df,2pd)以及6-311++g(3df,3pd)基组计算NaBr和NaF分子的几何结构、离解能以及谐振频率。计算值与实验值进行对照,结果发现在B3LYP/6-311g(2df,2pd)方法与基组计算得到NaBr分子平衡核间距Re=2.516 8Å,离解能De=3.729 7eV,与实验值Re=2.502 0Å,De=3.758 7eV吻合较好。在BP86/6-31g(3df,3pd)方法与基组计算得到NaF分子平衡核间距Re=1.929 0Å,离解能De=5.208 7eV,与实验值Re=1.925 9Å,De=5.363 1eV吻合较好。对NaBr和NaF分子分别选取B3LYP/6-311g(2df,2pd)、BP86/6-31g(3df,3pd)为最优方法与基组,在该方法下进行能量扫描,采用M-S势拟合,从而计算NaBr和NaF分子的力常数与光谱常数,对NaBr分子计算得到的谐振频率ωe为298.697 5cm-1、非谐振频率ωeXe为 1.354 2cm-1、刚性转动因子和非刚性转动因子Be和αe分别为0.149 5cm-1、0.961 5×10-3cm-1,对NaF分子计算得到的谐振频率ωe为543.686 6cm-1、非谐振频率ωeXe为3.434 4cm-1、刚性转动因子和非刚性转动因子Be和αe分别为0.435 5cm-1、0.430 6×10-2cm-1都与实验值吻合得很好。研究所采用的M-S势准确地拟合出基态的NaBr分子解析势能函数,这为进一步研究分子反应动力学提供了理论基础,所计算得到的NaBr和NaF分子光谱常数数据为NaBr和NaF分子在工业生产中的应用提供了理论依据。
参考文献:
[1] 朱正和,俞华根.分子结构与分子势能函数[M]. 北京:科学出版社,1997(8):1-10.
[2] 谢安东,朱正和.BF分子X1∑+,A1∑∏和B1∑+电子态的势能函数[J].化学学报, 2005,63(23):2126-2130.
[3] 赵俊,徐大海,程新路,等.KH(KD)分子基态(X1∑+)的结构与势能函数研究[J].化学学报,2010,68(6):461-465.
[4] 朱瑜,蒋刚,方芳,等.PdN、PdN2分子的结构与势能函数[J].物理化学学报,2006,22(5):538-541.
[5] 施毅敏,叶绍龙.NaBr的电子结构和光学性质的密度泛函研究[J].湖南师范大学学报(自然科学版),2009,32(4):32-36.
[6] 崔玉婷,朱永合.NaBr晶体的能带结构[J].重庆师范学院学报(自然科学版),1999,16(1):84-86.
[7] 詹勇军,王锋,袁玉全,等.NaF薄膜的结构与应力分析[J].重庆文理学院学报(自然科学版),2007,26(2):32-35.
[8] 朱正和,俞华根.原子分子反应静力学[M].北京:科学出版社,1996(4):30-78.
[9] HUBER K P,HERZBERG G.Molecular Spectra and Molecular Structure[M]. New York:Springer Science Business Media,1979(5):434-438.
文章编号:1004—5570(2016)01-0069-04
收稿日期:2015-10-18
基金项目:国家自然科学基金(No.10964002;No.10974139);贵州省科学技术基金(黔科合J字[2011]2110号);贵州省教育厅自然科学基金(黔教科20090041)和贵州师范大学博士科研基金
作者简介:赵淑令(1991-),女,硕士研究生,研究方向:原子与分子物理学,E-mail:shulingzhao521@foxmail.com. *通讯作者:沈光先(1972-),女,博士研究生,教授,研究方向:原子与分子物理学,E-mail: 362490965@qq.com.
中图分类号:O641.1
文献标识码:A
Potential energy functions and spectroscopic constants for the statesX1∑+of molecule NaX(X=Br, F)
ZHAO Shuling,LV Bing,SHEN Guangxian*
(School of Physics and Electronic Science, Guizhou Normal University, Guiyang, Guizhou 550001, China)
Abstract:The ground electronic state X1∑+ and dissociation limit of NaBr and NaF molecular have been correctly determined based on group theory atomic and molecular reaction statics. The equilibrium geometries, dissociation energy and harmonic frequencies of the ground state NaBr and NaF have been calculated using several methods in conjunction with different basis sets. For the NaBr molecular, It way chosen the B3LYP /6-311g (2df, 2pd) as the best method and basis sets. For the NaF molecular,It way chosen the BP86/6-31g (3df, 3pd) as the best method and basis sets. The whole potential curves for the ground electronic state have been scanned using the best method and basis set, the potential energy function of molecular have been obtained by least square fitting to the Murrell-Sorbie function. At the same time, the force constants and spectroscopic constants have been calculated. Comparing to the computation results, the present results were in good agreement with the experimental values.
Key words:NaBr; NaF; potential energy function; spectroscopic constants; Murrell-Sorbie
