肿瘤氧化还原代谢与干预
2016-07-10杨梦祺刘盼盼黄蓬
杨梦祺,刘盼盼,黄蓬
(中山大学肿瘤防治中心 华南肿瘤学国家重点实验室,广东 广州 510060)
肿瘤氧化还原代谢与干预
杨梦祺,刘盼盼,黄蓬Δ
(中山大学肿瘤防治中心 华南肿瘤学国家重点实验室,广东 广州 510060)
受肿瘤相关基因信号通路和代谢改变的影响,肿瘤细胞的氧化还原状态经常处于失衡状态,表现为活性氧(reactive oxygen species,ROS)水平异常增高及氧化胁迫(oxidative stress)。很多与肿瘤发生相关的信号通路都可直接或间接调节活性氧的代谢。异常升高的ROS在肿瘤细胞中的作用至今仍然备受争议,ROS一方面可促进细胞生长和肿瘤耐药,另一方面也能引起细胞的损伤甚至死亡。肿瘤细胞为适应高氧化应激而上调自身抗氧化能力,最终适应并维持存活。氧化应激的调节在肿瘤发生发展和对治疗的应答上都起着重要的作用。越来越多的研究提示通过靶向氧化还原调节机制的干预策略可以抑制肿瘤生长和消除肿瘤的耐药性。本文将阐述肿瘤细胞氧化还原代谢的改变和氧化应激及其调控机制,并讨论利用肿瘤这一生物化学特性进行干预的潜在治疗策略。
活性氧;肿瘤细胞;氧化应激;氧化还原适应
利用肿瘤细胞的生物学特征设计高效和高选择性药物及治疗策略一直是肿瘤研究的主要目标之一。近年来分子靶向药物的出现为提高肿瘤疗效和特异性带来了新希望,特异性针对正常细胞和肿瘤细胞之间基因差异的靶向治疗不仅能有效杀灭肿瘤细胞,而且对正常细胞的毒性也较小[1-2]。然而,获得性耐药和基因组的不稳定性使靶向治疗面临新的挑战[3-4]。肿瘤细胞内常有多个基因的改变,因此联合应用多种有针对性的特异性药物才有可能有效杀灭肿瘤细胞。然而,该联合策略的成功应用有赖于对肿瘤细胞赖以存活的多个关键基因组合的鉴定和认识,目前仍然需要多层次多角度深入的基础理论研究。由于大多数肿瘤细胞表现出有氧糖酵解的增加(Warburg效应)和高氧化应激状态[5],靶向这些特征性改变可望成为一种特异针对肿瘤细胞的治疗方法。
许多类型的肿瘤细胞都具有较正常细胞高的ROS水平[6]。细胞内的ROS主要来源于线粒体,其次是内质网和过氧化物酶体。另外,ROS也可来自细胞内其他涉及电子传递的生化反应及某些金属离子所介导的氧化还原反应。ROS主要包括两大类型,一是其外电子层含有不成对电子的自由基(free radical),另一类是外电子层不含不成对电子但化学性质活跃的非自由基活性氧(non-radical ROS)。ROS在生物机体中具有重要功能,包括氧化还原调节和生物信息传递等。适度的ROS增加可以促进细胞增殖和分化,但一旦过度升高则导致脂质、蛋白质和DNA的损伤[7]。维持ROS动态平衡是正常细胞存活和增殖的关键。细胞通过自身的活性氧代谢系统维持ROS的产生和清除的平衡而控制其合适的水平。利用基因技术和药物手段扰乱细胞的氧化还原平衡可导致肿瘤的发生和发展。Piskounova等[8]最近研究提示,增高的ROS对于促进和维持肿瘤细胞高转移表型可能具有关键作用。然而高氧化应激状态的肿瘤细胞更容易受外源性ROS诱导剂的损伤。因此,靶向氧化还原调节控制ROS水平可以作为一种有效的方式选择性的杀死肿瘤细胞[9]。
需要指出的是,肿瘤细胞中氧化还原代谢的改变非常复杂,应激反应和氧化还原调节机制涉及众多因素。在持续的氧化应激状态下,细胞可通过多种机制调节氧化应答功能,包括增强细胞内抗氧化系统的能力从而适应这种氧化胁迫[10],以及调节细胞存活过程中的转录因子和信号转导分子的活性和表达[11]。为了提高肿瘤治疗疗效和克服与氧化还原适应相关的耐药,必须阐明肿瘤细胞中复杂的氧化还原改变和潜在的机制。此外,由于肿瘤干细胞在肿瘤耐药和疾病复发中起着重要作用,检测肿瘤干细胞的活性氧代谢和氧化还原状态并制定相应的干预策略对肿瘤的有效治疗至关重要。
1 氧化应激在肿瘤中的作用和机制
大部分血液肿瘤和实体肿瘤,如慢性髓细胞白血病、慢性淋巴细胞白血病、肾癌、乳腺癌以及前列腺癌中都可以观察到ROS水平的明显升高[12-13]。此外,Valencia等[14]研究显示肿瘤相关成纤维细胞中ROS水平较正常成纤维细胞亦明显增高。虽然导致肿瘤细胞氧化应激的确切机制仍不很清楚,但目前认为以下内在和外在因素是肿瘤细胞高氧化应激的主要原因。
内在因素包括:①癌基因激活。早期研究发现MYC的异位高表达可以导致线粒体基因表达上调,增加ROS的产生[15]。最近对白血病干细胞的研究提示癌基因BCR-ABL的表达产物可诱导ROS产生[16]。肿瘤基因RAS的异常表达或突变可通过影响NADPH氧化酶而增加ROS的产生[17]。另一方面,MYC和RAS又可激活抗氧化机制而下调ROS[18],使肿瘤细胞达到新的氧化还原平衡;②能量代谢异常。多数肿瘤细胞具有较高的代谢活性,需要更多的三磷酸腺苷(ATP)作为能量以维持各项细胞活动。这高能量需求导致线粒体呼吸持续活跃,从而引起过量ROS的产生和累积[6];③线粒体结构功能异常。线粒体DNA编码电子传递链中的13种蛋白质,线粒体DNA的突变可导致呼吸链功能失调而造成电子异常渗漏。游离电子与分子氧反应产生超氧阴离子自由基(superoxide),随后可被转化为其他类型的ROS[19];④内质网功能异常。新生蛋白质在内质网(ER)中正确折叠才能保证其正常生理功能。错误折叠的蛋白质将引起内质网应激和未折叠蛋白质应答反应(unfolded protein response,UPR),导致ROS的产生和积聚[20]。另外,内质网应激诱导的钙释放和随后的线粒体内膜去极化亦可刺激线粒体ROS的产生[21];⑤抑癌基因失活。抑癌基因产物p53在感应和清除氧化损伤的核DNA和线粒体DNA,预防氧化基因突变和遗传物质不稳定中具有重要作用[22]。另外,p53作为转录因子可调控诸多抗氧化基因的表达[23]。因此,p53的缺失或突变可导致细胞的氧化还原失衡。
外在因素包括:炎性细胞因子、营养不平衡和乏氧环境[24]。肿瘤细胞增殖快,部分细胞常处于缺氧和营养不足的环境中,这将刺激线粒体产生ROS,异常增高的ROS又可反过来进一步激活缺氧诱导转录因子1(HIF1)。后者可促进血管生成、细胞存活和糖酵解[25]。
已有研究评估了细胞在不同环境下的ROS水平改变及其生理病理意义,旨在阐明ROS致瘤作用或抑瘤作用的环境条件。在适度的范围内,ROS可以刺激促有丝分裂原激活蛋白激酶(MAPK)和细胞外信号调节激酶(ERK)的磷酸化,细胞周期蛋白cyclin D1的表达和JUN氨基末端激酶(JNK)的活化,这些信号分子都与肿瘤的生长相关[26]。已有证据表明肿瘤细胞中一定水平的ROS增高有助于细胞增殖和存活、分化和迁移[27],并可促进免疫逃逸及耐药[28]。而当ROS严重升高而超过细胞抗氧化机制所能耐受的水平时,则可导致细胞损伤乃至死亡。需要回答的问题是,肿瘤细胞如何通过自身的调节机制维持一个比正常细胞高的ROS水平但又不至于影响细胞的各项生理功能,即肿瘤细胞的氧化应激适应?
2 肿瘤细胞对氧化应激的适应
早在20年前就有研究显示低剂量持续的外源性氧化剂预处理正常上皮细胞可赋予细胞抵抗后续高水平氧化应激的能力[29]。如图1所示,肿瘤细胞能够通过自身调控机制将ROS控制在合适的范围内。肾肿瘤细胞中磷酸戊糖代谢途径尤为活跃,以产生更多的还原当量NADPH。另外,多数肿瘤细胞的谷胱甘肽(GSH)合成能力增强。在恶性肿瘤细胞株和原代肿瘤组织中,活性氧清除酶超氧化物歧化酶(SOD1、SOD2和SOD3)、谷胱甘肽过氧化物酶、过氧化物酶体、硫氧还原蛋白(TRX)的水平均有不同程度的增高,还原型GSH和TRX也明显增多[30]。肿瘤细胞增强其氧化应激的能力主要是通过高表达负责清除ROS的酶系及产生GSH、NADPH、TRX等一系列的抗氧化分子。最近的研究也表明,某些诱导氧化应激的条件也会促进肿瘤细胞的抗氧化机制的激活[10]。

图1 细胞内氧化还原平衡示意图细胞内活性氧主要来源于线粒体电子传递链,内质网和NADPH氧化酶(NOX)复合物等。各种类型的活性氧在图中用红色标记出。过高的活性氧可导致蛋白质、脂质和DNA氧化损伤。为防止活性氧带来的损伤,细胞通过平衡活性氧的生成和消除而将ROS维持在一个适度的范围内。由于处于高度氧化应激状态,肿瘤细胞更加依赖抗氧化系统,包括氧化物酶体、谷胱甘肽过氧化物酶、过氧化氢酶、硫氧还原蛋白和谷氧还原蛋白等以对抗活性氧并减少其氧化损伤。一些药物如SSZ、BSO和AUR等可通过不同的机制干预细胞内活性氧清除系统Fig.1 Schematic illustration of cellular redox homeostasisCellular reactive oxygen species(ROS) are mainly generated from the mitochondrial electron transport chain, the endoplasmic reticulum(ER) system, and the NADPH oxidase(NOX) complex.Various types of ROS are highlighted in red color.The increases in ROS may lead to oxidative damage of protein, lipid, and DNA.To prevent the harmful effects of ROS, cells regulate ROS at a proper level by maintaining the balance between ROS generation and elimination.Due to high levels of intracellular ROS, cancer cells highly rely on antioxidant systems including peroxiredoxins, glutathione peroxidases, and catalase to counteract oxidative stress and minimize their damage.Several drugs such as sulfasalazine(SSZ), buthioninesulfoximine(BSO), and auranofin(AUR) could disrupt these ROS-scavenging systems
肿瘤细胞的氧化适应可能涉及多种信号途径以激活氧化还原敏感的转录因子,如NF-κB、NRF2、c-JUN和HIF-1等。这些转录因子的激活可促进超氧化物歧化酶、过氧化氢酶、硫氧还蛋白和谷胱甘肽等抗氧化系统的基因表达。例如,转录因子NRF2可激活多种能够降低活性氧的应激基因表达[31]。NRF2的获得性功能突变可赋予细胞高氧化应激能力和对一些抗肿瘤药物的抵抗能力,这有助于增强肿瘤细胞对某些化疗药物的抵抗[32]。NRF2还可以促进细胞的谷胱甘肽合成和利用,而谷胱甘肽具有促进烷化剂代谢和解毒的作用,还原型GSH可以逆转3溴丙酮酸(3-BrPA)对肿瘤的抑制作用[33]。因此,异常的药物代谢和细胞保护机制的激活可共同导致肿瘤细胞耐药。除了促进细胞的存活,这些氧化还原敏感的转录因子也可调节细胞增殖、细胞永生化、血管生成和肿瘤转移相关蛋白质的表达[34]。
另外,基因组的不稳定性也是肿瘤细胞氧化还原适应的重要机制之一。基因的不稳定性使其所编码蛋白质的功能改变或异常表达成为可能,并为细胞适应氧化应激环境及其在肿瘤微环境中获得生存优势提供了条件。
肿瘤细胞可通过氧化还原适应提高内源性的抗氧化系统的能力和激活细胞存活通路,从而将ROS维持在一个较稳定且可耐受的范围内(见图2)。上调的抗氧化系统,如GSH和NADPH等赋予细胞更强的抗氧化能力,增强DNA修复能力,从而减少了细胞凋亡。关于不同类型肿瘤细胞的氧化还原状态及其生理病理意义,目前尚缺乏系统研究。因此,人们对各种不同类型肿瘤细胞的氧化还原改变如何影响细胞表型的理解并不完整。对肿瘤细胞活性氧的代谢调控的深入研究,将为通过调控细胞的氧化还原状态而影响肿瘤细胞的行为提供潜在的治疗策略。
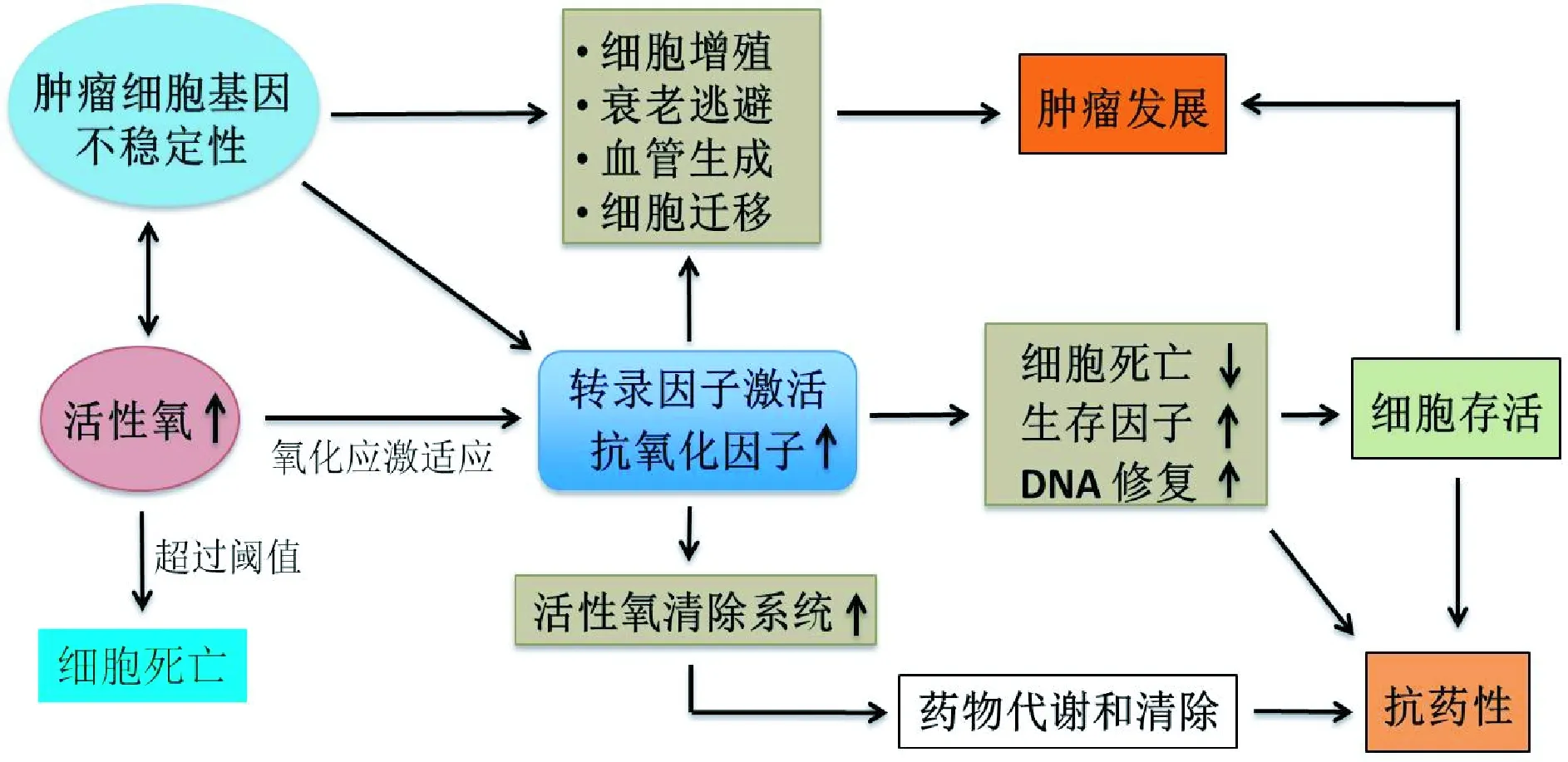
图2 肿瘤细胞中的氧化还原适应在持续的氧化应激下,肿瘤细胞激活氧化还原敏感性转录因子如NRF2等,导致抗活性氧系统(超氧化物歧化酶和谷胱甘肽等)的增加,存活因子表达升高和细胞死亡因子表达的抑制。这些转录因子可以调节与细胞增殖、衰老逃避、血管生成和肿瘤转移等相关基因的表达,促进肿瘤的发展。另一方面,ROS介导的DNA突变或缺失可促进基因组的不稳定性,这为异常基因表达提供了条件,有助于氧化应激的适应。此外,谷胱甘肽在应激适应过程中的增高能促进肿瘤细胞耐药。上述各种因素使肿瘤细胞在高氧化应激的情况下适应并存活Fig.2 Redox adaptation in cancer cellsIn response to persistent ROS stress, activation of redox-sensitive transcription factors, such as Nrf2, leads to an increase in the expression of ROS-scavenging molecules(such as superoxide dismutase and glutathione), elevation of survival factors, and anti-apoptotic factors.As these transcription factors also have roles in regulating the expression of genes that are responsible for proliferation, senescence evasion, angiogenesis and metastasis, these processes may promote cancer development.On the other hand, ROS-mediated DNA mutations promote genomic instability and this in turn contributes to stress adaptation.Furthermore, increased glutathione synthesis during adaptation can promote drug resistance.These together with enhanced cell survival may render cancer cells more resistant to chemotherapy.All these events enable cancer cells to survive with increased ROS and maintain their viability
3 靶向干预肿瘤细胞的氧化还原平衡状态
处于高氧化应激状态的肿瘤细胞相对于正常细胞更加依赖细胞的抗氧化系统,因此肿瘤细胞可能对活性氧诱导剂或抑制细胞抗氧化系统的化合物高度敏感。这一观点在二十年前就已经被提出,并为后来的研究结果所支持[35]。如图3所示,与正常细胞相比,肿瘤细胞可能因增高的ROS基础产率而对ROS的进一步诱导累积更为敏感。使用ROS诱导剂可使肿瘤细胞内ROS的含量升高,超过肿瘤细胞所能承受的阈值时,则导致肿瘤细胞的损伤和死亡。而正常细胞因自身ROS的基础水平较低,即使使用ROS诱导剂也不足以使其升高到致死阈值。尽管活性氧诱导剂在许多情况能有效杀灭肿瘤细胞,但亦有报道显示其疗效的局限性和肿瘤细胞对活性氧诱导剂的抵抗性[35]。因此,理解不同氧化还原调控策略的利弊以及肿瘤细胞内在的氧化应激状态和氧化还原适应机制对设计有效的干预策略十分重要。目前干预肿瘤细胞氧化还原状态主要有以下4种常用策略(见图4)。

图3 活性氧介导选择性治疗的生物学基础在生理情况下,正常细胞维持较低的基础活性氧水平并能够耐受一定程度的氧化应激。而肿瘤细胞处于较高的氧化应激状态但其活性氧水平尚未达到损伤细胞的水平。因此,肿瘤细胞对抗氧化系统更加依赖,对内外源性的氧化刺激更加敏感。进一步升高肿瘤细胞的ROS可以使其超过细胞所能承受的阈值并最终导致肿瘤细胞的死亡。而在同等程度的氧化刺激下,正常细胞较低的基础ROS即使继续升高也达不到细胞损伤死亡的阈值。正常细胞与肿瘤细胞之间这一差异可为活性氧介导选择性抗肿瘤治疗提供生化依据Fig.3 A biological basis for ROS-mediated therapeutic selectivity Under physiological conditions, normal cells maintain a low basal ROS level and can tolerate a certain level of further ROS stress owing to their low ROS output and intact antioxidant capacity, which together prevent the ROS level from reaching the cell-death threshold under ROS stimulation.In contract, cancer cells have higher ROS output just below the threshold level, and would be more dependent on the antioxidant system and more vulnerable to further oxidative stress induced by ROS stimuli.A further increase of ROS stress in cancer cells is likely to cause a severe elevation of ROS above the threshold level, leading to cell death.This might provide a biochemical basis to design selective therapeutic strategies using ROS-mediated mechanisms

图4 通过活性氧介导机制靶向杀灭肿瘤细胞细胞内活性氧水平是由活性氧产生和消除所决定的。适度增加活性氧可以通过氧化还原适应机制来调节并不对细胞产生毒性作用。然而,显著增加ROS产生和/或抑制其消除能诱导肿瘤细胞内ROS显著积累,超过细胞的应激适应能力时将导致氧化损伤和细胞死亡。图中标出4种能导致活性氧严重积累的潜在抗肿瘤策略Fig.4 Targeting cancer cells through ROS-mediated mechanismsCellular ROS level is determined by ROS generation and elimination.A moderate ROS increase can by balanced by redox adaptation mechanisms and thus avoid cytotoxicity.However, a severe increase in ROS generation and/or inhibit ROS elimination can induce significant accumulation of ROS in cancer cells,which cannot be reversed by redox adaptation mechanisms.This will lead oxidative damage and cell death.Four potential intervention strategies to induce severe ROS accumulation as potential therapeutic approaches are indicated in the figure
3.1 促进ROS的产生 尽管肿瘤细胞具有较强的抗氧化系统,其ROS仍然维持在比正常细胞更高的水平。因此,肿瘤细胞可能比正常细胞对ROS的进一步累积更敏感,这就为选择性杀灭肿瘤细胞提供了一个治疗窗。细胞的氧化还原状态由活性氧的生成和消除速率的精细调节以维持平衡。增加ROS产生或减少ROS清除会破坏氧化还原平衡,导致ROS水平升高,当ROS超过细胞耐受的阈值时就会导致细胞损伤乃至细胞死亡。很多药物都可以升高ROS水平进而选择性杀伤肿瘤细胞。治疗白血病的有效药物三氧化二砷可通过干预呼吸链的功能增加ROS的产生[36]。紫杉烷类(紫杉醇和多西他赛)、长春花生物碱(长春新碱和长春碱)和抗代谢物(抗叶酸)能促进细胞色素c从线粒体释放进而诱导细胞死亡,这一过程也干扰了电子传递链,造成超氧自由基的产生[37]。铂类药物(顺铂、卡铂和奥沙利铂)和蒽环类(阿霉素、表柔比星和柔红霉素)也通过不同的机制诱导产生大量的ROS[38]。有报道认为,临床广泛应用的5-氟尿嘧啶(5-FU)可通过p53依赖途径诱导线粒体活性氧的生成[39]。此外,放射治疗可导致ROS的显著增加[40]。内质网应激亦可诱导氧化应激而杀伤肿瘤细胞,蛋白酶抑制剂硼替佐米正是通过此机制诱导ROS的异常增高[41]。
3.2 干扰活性氧代谢 细胞内ROS的消除依靠多个抗氧化系统,包括还原型谷胱甘肽(GSH)系统,硫氧还原蛋白系统和过氧化氢酶等。可以通过靶向这些抗氧化系统中的关键酶,进而抑制细胞的抗氧化能力和消除氧化应答所造成的抗药性。目前作用于细胞内谷胱甘肽的药物有丁硫氨酸砜(BSO)、异硫氰酸酯和氮丙啶衍生物等。BSO是谷胱甘肽合成的限速酶谷氨酰半胱氨酸合成酶(γ-GCS)的抑制剂,该化合物已被证明在多种类型的肿瘤细胞中可导致GSH减少进而体现其抗癌活性[42]。异硫氰酸酯能与GSH结合而导致GSH的快速耗尽。此外,胱氨酸/谷氨酸转运蛋白抑制剂,如柳氮磺胺吡啶,可通过抑制谷胱甘肽合成中的限速底物半胱氨酸的前体胱氨酸的摄入而导致细胞内GSH的减少[43]。对于硫氧还原蛋白系统,1-甲基丙基2-咪唑基二硫化物(PX-12)是硫氧还原蛋白1(TRX-1)的抑制剂,在体内具有抗肿瘤活性[44]。然而,细胞质中的抗氧化剂并不能有效清除线粒体中的ROS,有研究表明在动物模型中使用线粒体特异性的抗氧化剂可以显著减少成瘤率[45]。
考虑到氧化应激在肿瘤发生发展过程中的重要作用,通过干预氧化还原代谢消除氧化应激反应已被认为是预防和治疗肿瘤的重要策略。然而,氧化还原干预在肿瘤治疗中的作用非常复杂,因而存在不少争议。曾有报道显示,抗氧化剂N-乙酰葡糖胺(NAC)可以抑制淋巴瘤在小鼠体内的生长[46],但后来的研究又发现NAC可增加小鼠黑色素瘤的转移病灶[8]。大多数抗氧化剂在临床试验中并未显示出有效的治疗效果。一个可能的解释是,细胞为了存活启动了有效的氧化还原调控,即使外源性给予抗氧化剂,对体内细胞的氧化还原平衡状态影响也相对有限。需要特别指出的是,因为多数肿瘤细胞处于高氧化应激状态,给予抗氧化剂可能利于肿瘤的细胞存活并增强其抗药性。因此,这方面的临床研究必须十分慎重。
3.3 克服与氧化应激相关的耐药性 鉴于正常细胞与恶性细胞之间ROS含量的差异,给予外源性活性氧诱导剂可优先杀伤肿瘤细胞。然而,肿瘤细胞在持续高氧化应激的环境下可能上调细胞的抗氧化系统,表现在上调的细胞内GSH含量,增高某些抗氧化酶的表达等。例如,耐药的白血病细胞HL-60可抵抗过氧化氢的细胞毒作用,这很可能是由于该细胞高表达过氧化氢酶所致[47]。肿瘤细胞对三氧化二砷的抵抗与细胞内HMOX1、SOD1和GSH的上调有关[48]。同样,研究表明ROS诱导剂,如紫杉醇,阿霉素或铂类药物的耐药与细胞抗氧化能力的升高相关[49]。因此,耐药与肿瘤细胞对内源性氧化应激的适应有密切关系。
基于上述原因,消除肿瘤细胞对氧化应激的适应机制是克服肿瘤耐药的重要措施。一种策略是靶向干预控制ROS和氧化还原敏感性的关键分子。谷胱甘肽,硫氧还原蛋白和过氧化物酶被认为是氧化还原干预的潜在靶标。以往研究显示,天然化合物苯乙基异硫氰酸酯(PEITC)能够优先杀伤RAS诱导的恶性转化的卵巢细胞和来自患者的原代白血病细胞,并有效克服其对格列卫的耐药性。PEITC可使肿瘤细胞内的谷胱甘肽迅速耗尽,有效破坏氧化应激的适应机制并导致对氧化还原敏感的MCL-1等生存蛋白的降解[50]。前已述及,单独使用活性氧诱导剂对肿瘤可能疗效不佳,故更好的策略可能是活性氧诱导剂与能够消除氧化应激适应机制的化合物联用。最近的研究显示,PEITC和其他抗肿瘤药物联合可产生更强的抗肿瘤能力[51]。随着该领域研究的进展,越来越多的新型化合物能有效的破坏肿瘤细胞内的氧化还原机制从而实现有效杀灭肿瘤细胞并克服肿瘤耐药。
3.4 氧化还原干预的联用药物策略 在肿瘤治疗中,单独使用某一特定的药物较容易出现耐药,疗效也相对较差。因此,临床上大多采用联合用药的治疗方案。由于细胞的氧化还原平衡状态由ROS的产生和活性氧的消除率2个方面所决定,这为联合用药干预肿瘤细胞的氧化还原平衡提供了合适的生化基础。上述的联合ROS诱导剂和抑制细胞抗氧化能力的化合物PEITC就是该联合用药方案的一个例子。活性氧诱导剂三氧化二砷和超氧化物歧化酶抑制剂联合应用对原发性慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)有显著疗效,对CLL细胞的毒性明显增强[52]。联合三氧化二砷和消耗GSH的抗坏血酸在治疗复发或难治的多发性骨髓瘤中也有明显治疗效果[53]。在鼻咽癌中使用硼替唑米联合SAHA(辛二酰苯胺异羟肟酸)具有协同作用,导致ROS介导的细胞死亡[54]。最近的研究显示,联合靶向线粒体呼吸链的药物和抑制清除线粒体ROS的药物可以阻止原发性肝细胞肿瘤在小鼠体内的转移[45]。
此外,研究发现联合抑制GSH代谢和硫氧还蛋白代谢的药物可增强肿瘤细胞对17-烯丙基氨基-17-去甲氧基格尔德霉素(17-AAG)的治疗敏感性[55]。热休克蛋白90(HSP90)的上调是肿瘤细胞重要的保护机制,而17-AAG能结合HSP90并对其产生抑制作用。HSP90抑制剂与mTOR抑制剂雷帕霉素联合可导致内质网应激和线粒体损伤,从而增强由活性氧介导的杀灭肿瘤细胞的活性,并明显降低肿瘤在小鼠体内的生长[56]。因此,HSP90抑制剂与诱导氧化应激的药物联合能有效杀灭肿瘤细胞。另外,有研究显示聚腺苷二磷酸-核糖聚合酶(PARP)抑制剂,如veliparib或olaparib与卡铂联用对多种不同类型的肿瘤都有较好的治疗效果[57]。放射治疗和传统的细胞毒性抗癌药物大都可以直接或间接地增加肿瘤细胞中ROS水平,因此联合放疗或传统化疗与抑制抗氧化系统的药物有望取得治疗肿瘤的良好疗效。
4 肿瘤干细胞的氧化还原状态
化疗或放疗后肿瘤复发的重要原因之一是未能彻底杀灭具有高度抗药性的肿瘤干细胞[58]。考虑到氧化还原适应在肿瘤耐药中的重要作用,因此推测肿瘤干细胞耐药也与氧化还原调控机制密切相关。与一般肿瘤细胞相比,肿瘤干细胞的ROS水平较低,这有利于肿瘤干细胞维持自我更新能力和干性。肿瘤干细胞有较低的ROS水平的主要原因之一是抗氧化物质GSH的合成增高和磷酸戊糖途径的高活性,后者产生较多还原物质NADPH。另外,肿瘤干细胞的线粒体活性氧的产生可能下降(见图5)。有研究发现CD44+的胃肠肿瘤干细胞通过激活胱氨酸谷氨酸转运载体而增加GSH的合成。肿瘤干细胞中低水平的ROS有助于其抵抗放射治疗,因为放射线主要通过诱导自由基的产生而发挥其对细胞的毒性作用,而肿瘤干细胞中的高GSH和低ROS水平不利于放射线对细胞的损伤作用[59]。另外,肿瘤干细胞在体内常处于相对缺氧的环境中,这些缺氧的环境可能限制其分化和维持其干性[60]。缺氧可能会限制内源性活性氧的产生,不利于放射线对肿瘤干细胞的杀灭。残留的肿瘤干细胞会导致日后肿瘤的复发。

图5 肿瘤干细胞内低水平ROS的可能机制线粒体呼吸下调导致ROS生成减少,磷酸戊糖途径代谢活跃使NADPH生成增加以及增高的GSH合成能力等因素使肿瘤干细胞内的ROS水平降低。这些因素可促进肿瘤干细胞细胞存活和耐药性Fig.5 Possible mechanisms for maintaining a low level of ROS in cancer stem cellsDecreased ROS production from low mitochondrial respiration, increased NADPH production through pentose phosphate metabolism, and increased GSH synthesis together result in a low level of ROS in cancer stem cells.This can promote cancer stem cell survival and resistance to therapy
考虑到肿瘤干细胞的生物学特性,可以推测这群特殊的细胞能够通过较高的抗氧化能力将ROS维持在一个较低的范围内以保持其干性和肿瘤形成的能力。肿瘤干细胞的存活和耐药可能归因于高度上调的抗氧化机制。在这个重要的研究领域中,首先确定肿瘤干细胞相比于正常干细胞和一般肿瘤细胞的氧化还原状态是很重要的。明确肿瘤干细胞中维持氧化还原平衡的关键分子机制是设计有效杀灭这些细胞以彻底清除肿瘤的关键。破坏肿瘤干细胞的抗氧化系统是否能够增加对其他抗肿瘤药物的敏感性?外源性的活性氧应激是否能够导致肿瘤干细胞失去其干性?氧化还原调节策略联合传统抗癌药物是否能够更有效杀灭肿瘤干细胞?这些问题尚需进一步深入研究。
5 结论和展望
肿瘤细胞的高ROS水平可能促进细胞增殖,而对高氧化应激的适应可能促进细胞存活和抗药性。这2种现象体现了氧化应激在肿瘤发生发展及耐药中的关键作用。通过氧化还原调节策略靶向干预肿瘤细胞的这些生化特征可望提高抗肿瘤的选择性和克服抗药性。氧化应激一方面促进肿瘤发展,另一方面导致氧化损伤,这为肿瘤治疗提供了2种可能的干预策略。一种策略是通过抗氧化剂来增加ROS清除的能力,以期抑制肿瘤生长。然而,大规模的前瞻性随机临床试验发现在长期服用抗氧化剂维生素E的健康男性人群中前列腺癌的发生率升高,这提示抗氧化剂可能促进某些肿瘤的发生[7]。也有实验数据显示抗氧化剂NAC和维生素E通过减少ROS从而降低DNA损伤和p53的表达,有利于肿瘤细胞的增殖。此外,由于非干细胞在一定条件下可以向干细胞转化,如果低水平的ROS利于维持细胞的干性,那么抗氧化剂将有可能促进肿瘤干细胞的生成[59]。一些研究者推测,补充抗氧化剂可用于预防肿瘤或者增强化疗和放疗疗效。然而,最近的研究亦显示出相反的结果,提示抗氧化剂能够促进肿瘤细胞的存活和增殖。
另一种治疗策略是用具有氧化作用或能够抑制抗氧化系统的药物来进一步升高ROS从而杀灭肿瘤细胞。在实验模型中,ROS诱导剂通过升高胞内的ROS,使这一氧化应激超过细胞抗氧化系统所能承受的阈值而杀灭肿瘤细胞[61]。GSH作为细胞内主要的抗氧化剂,干预其代谢可望成为抗肿瘤药物的重要靶标。然而,其他具有抗氧化能力的酶对肿瘤细胞的存活也是至关重要的,是潜在的治疗靶标。进一步探究细胞内抗氧化分子的代谢和调控将利于设计更有效的抗肿瘤治疗策略。联合使用ROS诱导剂和抑制ROS清除的药物能引起肿瘤细胞内ROS严重升高,可能是提高抗肿瘤作用的有效策略。然而这一策略也可能促进正常细胞突变。因此,通过调节ROS治疗肿瘤是否获益目前仍有一定的争议。
近年来大量的研究表明改变细胞氧化还原代谢在不同类型肿瘤的发展中起着至关重要的作用。对高氧化应激的适应在一定程度上解释了肿瘤细胞在持续内源性高氧化状态下的存活和抗药性。肿瘤细胞普遍具有增强的抗氧化能力,这与放疗化疗抵抗相关。因此,通过药物消除这一适应机制并联合传统化放疗可望提高疗效和克服肿瘤细胞的抗药性。由于肿瘤干细胞在肿瘤的发生发展及耐药性过程中起关键作用,通过氧化还原调节的手段消除肿瘤干细胞在肿瘤治疗中具有重要意义。目前对肿瘤干细胞的的氧化还原状态及其调控的认识尚属初期阶段,有许多不明确及争议之处。随着新技术的发展,特别是代谢组学分析技术与活性氧实时检测及细胞内活性氧的定位分析技术将大幅度推进对肿瘤氧化应激和代谢适应及其调控机制的研究,并据此设计出更有效的干预策略,以期提高肿瘤疗效。
[1] Tolaney SM,Barry WT,Dang CT,et al.Adjuvant paclitaxel and trastuzumab for node-negative,her2-positive breast cancer[J].New Engl J Med,2015,372(2):134-141.
[2]Hunger SP,Mullighan CG.Acute lymphoblastic leukemia in children[J].New Engl J Med,2015,373(16):1541-1552.
[3]McDermott U,Downing JR,Stratton MR.Genomics and the continuum of cancer care[J].New Engl J Med,2011,364(4):340-350.
[4]Bosbach B,Deshpande S,Rossi F,et al.Imatinib resistance and microcytic erythrocytosis in a kitv558Δ;t669I/+ gatekeeper-mutant mouse model of gastrointestinal stromal tumor[J].Proc Natl Acad Sci U S A,2012,109(34):E2276-2283.
[5]Pavlova NN,Thompson CB.The emerging hallmarks of cancer metabolism[J].Cell Metab,2016,23(1):27-47.
[6]Cairns RA,Harris IS,Mak TW.Regulation of cancer cell metabolism[J].Nat Rev Cancer,2011,11(2):85-95.
[7]Schumacker PT.Reactive oxygen species in cancer: a dance with the devil[J].Cancer Cell,2015,27(2):156-157.
[8]Piskounova E,Agathocleous M,Murphy MM,et al.Oxidative stress inhibits distant metastasis by human melanoma cells[J].Nature,2015,527(7577):186-191.
[9]Schumacker PT.Reactive oxygen species in cancer cells: Live by the sword,die by the sword[J].Cancer Cell,2006,10(3):175-176.
[10]Diehn M,Cho RW,Lobo NA,et al.Association of reactive oxygen species levels and radioresistance in cancer stem cells[J].Nature,2009,458(7239):780-783.
[11]Hossain MS,Dietz KJ.Tuning of redox regulatory mechanisms,reactive oxygen species and redox homeostasis under salinity stress[J].FrontPlant Sci,2016,7:548.
[12]Singh AK,Awasthi D,Dubey M,et al.High oxidative stress adversely affects nfkappab mediated induction of inducible nitric oxide synthase in human neutrophils: Implications in chronic myeloid leukemia[J].Nitric Oxide,2016,58:28-41.
[13]Cancer Genome Atlas Research N.Comprehensive molecular characterization of clear cell renal cell carcinoma[J].Nature,2013,499(7456):43-49.
[14]Valencia T,Kim JY,Abu-Baker S,et al.Metabolic reprogramming of stromal fibroblasts through p62-mtorc1 signaling promotes inflammation and tumorigenesis[J].Cancer Cell,2014,26(1):121-135.
[15]Tanaka H,Matsumura I,Ezoe S,et al.E2f1 and c-myc potentiate apoptosis through inhibition of nf-kappab activity that facilitates mnsod-mediated ros elimination[J].MolCell,2002,9(5):1017-1029.
[16]Capala ME,Pruis M,Vellenga E,et al.Depletion of sam50 specifically targets bcr-abl-expressing leukemic stem and progenitor cells by interfering with mitochondrial functions[J].Stem Cells Dev,2016,25(5):427-437.
[17]Keeling MJ,Danon L,Vernon MC,et al.Individual identity and movement networks for disease metapopulations[J].Proc Natl Acad Sci U S A,2010,107(19):8866-8870.
[18]Vafa O,Wade M,Kern S,et al.C-myc can induce DNA damage,increase reactive oxygen species,and mitigate p53 function: A mechanism for oncogene-induced genetic instability[J].Mol Cell,2002,9(5):1031-1044.
[19]Brandon M,Baldi P,Wallace DC.Mitochondrial mutations in cancer[J].Oncogene,2006,25(34):4647-4662.
[20]Pagliassotti MJ,Kim PY,Estrada AL,et al.Endoplasmic reticulum stress in obesity and obesity-related disorders: an expanded view[J].Metabolism,2016,65(9):1238-1246.
[21]Bravo R,Gutierrez T,Paredes F,et al.Endoplasmic reticulum: Er stress regulates mitochondrial bioenergetics[J].Int J Biochem Cell B,2012,44(1):16-20.
[22]Achanta G,Sasaki R,Feng L,et al.Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA pol gamma[J].The EMBO journal,2005,24(19):3482-3492.
[23]Sablina AA,Budanov AV,Ilyinskaya GV,et al.The antioxidant function of the p53 tumor suppressor[J].Nat Med,2005,11(12):1306-1313.
[24]Vyas S,Zaganjor E,Haigis MC.Mitochondria and cancer[J].Cell,2016,166(3):555-566.
[25]Semenza GL.Oxygen sensing,homeostasis,and disease[J].New Engl J Med,2011,365(6):537-547.
[26]Song JS,Kim EK,Choi YW,et al.Hepatocyte-protective effect of nectandrin b,a nutmeg lignan,against oxidative stress: Role of nrf2 activation through erk phosphorylation and ampk-dependent inhibition of gsk-3beta[J].Toxicol Appl Pharm,2016,307:138-149.
[27]Rhee SG.Cell signaling.H2o2,a necessary evil for cell signaling[J].Science,2006,312(5782):1882-1883.
[28]Zhong Y,Zhang F,Sun Z,et al.Drug resistance associates with activation of nrf2 in mcf-7/dox cells,and wogonin reverses it by down-regulating nrf2-mediated cellular defense response[J].Mol Carcinogen,2013,52(10):824-834.
[29]Choi J,Liu RM,Forman HJ.Adaptation to oxidative stress: Quinone-mediated protection of signaling in rat lung epithelial l2 cells[J].Biochem Pharmacol,1997,53(7):987-993.
[30]Liu HW,Zhu X,Zhang J,et al.A red emitting two-photon fluorescent probe for dynamic imaging of redox balance meditated by a superoxide anion and gsh in living cells and tissues[J].Analyst,2016.
[31]Kato J,Lanier-Smith KL,Currie MG.Cyclic gmp down-regulates atrial natriuretic peptide receptors on cultured vascular endothelial cells[J].J Biol Chem,1991,266(22):14681-14685.
[32]Sullivan LB,Martinez-Garcia E,Nguyen H,et al.The proto-oncometabolite fumarate binds glutathione to amplify ros-dependent signaling[J].Mol cell,2013,51(2):236-248.
[33]Rodrigues-Ferreira C,da Silva AP,Galina A.Effect of the antitumoral alkylating agent 3-bromopyruvate on mitochondrial respiration: Role of mitochondrially bound hexokinase[J].J Bioenerg Biomembr,2012,44(1):39-49.
[34]Chen EI,Hewel J,Krueger JS,et al.Adaptation of energy metabolism in breast cancer brain metastases[J].Cancer Res,2007,67(4):1472-1486.
[35]Hileman EO,Liu J,Albitar M,et al.Intrinsic oxidative stress in cancer cells: A biochemical basis for therapeutic selectivity[J].Cancer Chemo Pharm,2004,53(3):209-219.
[36]You BR,Park WH.Arsenic trioxide induces human pulmonary fibroblast cell death via increasing ros levels and gsh depletion[J].Oncol Rep,2012,28(2):749-757.
[37]Kaufmann SH,Earnshaw WC.Induction of apoptosis by cancer chemotherapy[J].Expe Cell Res,2000,256(1):42-49.
[38]Conklin KA.Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness[J].Integr Cancer Ther,2004,3(4):294-300.
[39]Hwang PM,Bunz F,Yu J,et al.Ferredoxin reductase affects p53-dependent,5-fluorouracil-induced apoptosis in colorectal cancer cells[J].Nat Med,2001,7(10):1111-1117.
[40]Kobashigawa S,Kashino G,Suzuki K,et al.Ionizing radiation-induced cell death is partly caused by increase of mitochondrial reactive oxygen species in normal human fibroblast cells[J].Radiat Res,2015,183(4):455-464.
[41]Kardosh A,Golden EB,Pyrko P,et al.Aggravated endoplasmic reticulum stress as a basis for enhanced glioblastoma cell killing by bortezomib in combination with celecoxib or its non-coxib analogue,2,5-dimethyl-celecoxib[J].Cancer Res,2008,68(3):843-851.
[42]Hass C,Belz K,Schoeneberger H,et al.Sensitization of acute lymphoblastic leukemia cells for lcl161-induced cell death by targeting redox homeostasis[J].Biochem Pharmacol,2016,105:14-22.
[43]Lewerenz J,Hewett SJ,Huang Y,et al.The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities[J].Antioxid Redox Sign,2013,18(5):522-555.
[44]Welsh SJ,Williams RR,Birmingham A,et al.The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation[J].Mol Cancer Ther,2003,2(3):235-243.
[45]Porporato PE,Payen VL,Perez-Escuredo J,et al.A mitochondrial switch promotes tumor metastasis[J].Cell Rep,2014,8(3):754-766.
[46]Gao P,Zhang H,Dinavahi R,et al.Hif-dependent antitumorigenic effect of antioxidants in vivo[J].Cancer cell,2007,12(3):230-238.
[47]Suzuki T,Spitz DR,Gandhi P,et al.Mammalian resistance to oxidative stress: A comparative analysis[J].Gene expression,2002,10(4):179-191.[48]Nishimoto S,Suzuki T,Koike S,et al.Nrf2 activation ameliorates cytotoxic effects of arsenic trioxide in acute promyelocytic leukemia cells through increased glutathione levels and arsenic efflux from cells[J].Toxicol Appl Pharm,2016,305:161-168.
[49]Ramanathan B,Jan KY,Chen CH,et al.Resistance to paclitaxel is proportional to cellular total antioxidant capacity[J].Cancer Res,2005,65(18):8455-8460.
[50]Zhang H,Trachootham D,Lu W,et al.Effective killing of gleevec-resistant cml cells with t315i mutation by a natural compound peitc through redox-mediated mechanism[J].Leukemia,2008,22(6):1191-1199.
[51]Kasukabe T,Honma Y,Okabe-Kado J,et al.Combined treatment with cotylenin a and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells[J].Oncol Rep,2016,36(2):968-976.
[52]Zhou Y,Hileman EO,Plunkett W,et al.Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ros-generating anticancer agents[J].Blood,2003,101(10):4098-4104.
[53]Dalton WS.Targeting the mitochondria: An exciting new approach to myeloma therapy.Commentary re: N.J.Bahlis et al.,Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma.Clin.Cancer Res.,8: 3658-3668,2002[J].Clin Cancer Res,2002,8(12):3643-3645.
[54]Hui KF,Lam BH,Ho DN,et al.Bortezomib and saha synergistically induce ros-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of epstein-barr virus[J].Mol Cancer Ther,2013,12(5):747-758.
[55]Scarbrough PM,Mapuskar KA,Mattson DM,et al.Simultaneous inhibition of glutathione- and thioredoxin-dependent metabolism is necessary to potentiate 17aag-induced cancer cell killing via oxidative stress[J].Free Radical Bio Med,2012,52(2):436-443.
[56]De Raedt T,Walton Z,Yecies JL,et al.Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors[J].Cancer cell,2011,20(3):400-413.
[57]Clark CC,Weitzel JN,O'Connor TR.Enhancement of synthetic lethality via combinations of abt-888,a parp inhibitor,and carboplatin in vitro and in vivo using brca1 and brca2 isogenic models[J].Mol Cancer Ther,2012,11(9):1948-1958.
[58]Yoshida GJ,Saya H.Therapeutic strategies targeting cancer stem cells[J].Cancer Sci,2016,107(1):5-11.
[59]Schieber MS,Chandel NS.Ros links glucose metabolism to breast cancer stem cell and emt phenotype[J].Cancer cell,2013,23(3):265-267.[60]Rivera A,Maxwell SA.The p53-induced gene-6(proline oxidase) mediates apoptosis through a calcineurin-dependent pathway[J].J Biol Chem,2005,280(32):29346-29354.
[61]Yu T,Sheu SS,Robotham JL,et al.Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species[J].Cardiovasc Res,2008,79(2):341-351.
(编校:吴茜)
作 者 简 介

黄蓬,教授,博士生导师。毕业于美国德州大学休斯顿生物医学研究生院,获博士学位。2007年成为美国德州大学M.D.Anderson癌症中心终身教授;2008年教育部长江学者讲座教授,2010年国家“千人计划”特聘专家,任中山大学肿瘤防治中心教授。主要研究方向为肿瘤代谢,肿瘤细胞线粒体功能改变,活性氧应激调控及代谢靶向抗癌药物。代表性论文发表于Nature,CancerCell,NatureCellBiology,JNCI,NatureRevDrugDiscovery等学术刊物,现为MolecularCancer副主编及多家专业杂志的编委。
ROS-mediated mechanisms: an anticancer strategy
YANG Meng-qi, LIU Pan-pan, HUANG PengΔ
(Department of Experimental Research, SunYat-sen University Cancer Center, Guangzhou 510060, China)
Under the influence of oncogenic signals and abnormal metabolism, the redox status of cancer cells often differs from that of the normal cells, manifesting as elevated generation of reactive oxygen species(ROS) and oxidative stress.Many signaling pathways involved in tumorigenesis can directly or indirectly regulate ROS metabolism.Currently, the biological significance of increased ROS in cancer cells is still somewhat controversial.ROS on the one hand can promote cancer development and drug resistance, and on the other hand can also cause cell injury and even cell death.To maintain cell viability and proliferation, cancer cells usually up-regulate their antioxidant capacity in adaptation to the intrinsic oxidative stress.Such adaptive mechanisms to oxidative stress are highly important in both cancer development and also play a major role in cancer cell response to therapy.Compelling evidences from recent studies have suggested that targeting the redox regulation mechanisms through proper intervention strategies may have significant therapeutic implications in cancer treatment.This article will focus on alterations of redox status in cancer cells, their adaptation to oxidative stress, and the underlying mechanisms.Potential therapeutic strategies based on such biochemical characteristics of cancer cells will also be discussed.
reactiveoxygen species; cancer cells; ROS stress; redox adaptation
10.3969/j.issn.1005-1678.2016.09.004
国家自然科学基金(81430060;81502573);广东省自然科学基金(2014A030310421)
杨梦祺,女,博士在读,E-mail:yangmq@sysucc.org.cn;黄蓬,通信作者,男,教授,博士生导师,研究方向:肿瘤代谢,肿瘤细胞线粒体功能改变,活性氧应激调控及代谢靶向抗癌药物,E-mail:huangpeng@sysucc.org.cn。
R73
A
