Fabrication of multiwalled carbon nanotube-surfactant modified sensor for the direct determination of toxic drug 4-aminoantipyrine☆
2015-11-17JayantGowdaArunkumarBuddanavarSharanappaNandibewoor
Jayant I.Gowda,Arunkumar T.Buddanavar,Sharanappa T.Nandibewoor
P.G.Department of Studies in Chemistry,Karnatak University,Dharwad 580003,India
Original Article
Fabrication of multiwalled carbon nanotube-surfactant modified sensor for the direct determination of toxic drug 4-aminoantipyrine☆
Jayant I.Gowda,Arunkumar T.Buddanavar,Sharanappa T.Nandibewoor*
P.G.Department of Studies in Chemistry,Karnatak University,Dharwad 580003,India
A R T I C L E I N F O
Article history:
7 January 2015
Accepted 7 January 2015
Available online 17 January 2015
Voltammetry
Modified electrode
Diffusion controlled
4-aminoantipyrine
Pharmacokinetic study
A multi-walled carbon nanotube(MWCNT)-cetyltrimethylammonium bromide(CTAB)surfactant composite modified glassy carbon electrode(GCE)was developed as a novel system for the determination of 4-aminoantipyrine(AAP).The oxidation process was irreversible over the pH range studied and exhibited a diffusion controlled behavior.All experimental parameters were optimized.The combination of MWCNT-CTAB endows the biosensor with large surface area,good biological compatibility,electricity and stability,high selectivity and sensitivity.MWCNT-CTAB/GCE electrode gave a linear response for AAP from 5.0×10-9to 4.0×10-8M with a detection limit of 1.63×10-10M.The modified electrode showed good selectivity against interfering species and also exhibited good reproducibility.The present electrochemical sensor based on the MWCNT-CTAB/GCE electrode was applied to the determination of AAP in real samples.
©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Electrochemistry has many advantages,making it an appealing choice for pharmaceutical analysis[1,2].Electrochemistry has always provided analytical techniques characterized by instrumental simplicity,moderate cost,and portability.These techniques have introduced the most promising methods for specific applications[3,4].Due to similarity in the electrochemical and biological reactions,it can be assumed that the oxidation/reduction mechanisms taking place at the electrode and in the body share similar principles.Biologically important molecules can be investigated electroanalytically by voltammetry in order to determine the molecule in different ways.Additional applications of electrochemistry include the determination of electrode mechanisms.The redox properties of drugs can give us insights into their metabolic fate in in vivo redox processes or pharmacological activity[5].
Further,the electroanalytical techniques have been shown to be excellent for the determination of pharmaceutical compounds in different matrices.Many of the active constituents of formulations,in contrast to excipients,can be readily oxidized.The selectivity of this method is normally excellent because the analyte can be readily identified by its voltammetric peak potential. Experimental electrochemical techniques have advantages in simplicity,cost,and analysis time,compared to other techniques in the field of drug analysis.The use of various electrodes,viz. mercury[6],solids[7,8],and modified electrodes[9-18],for electroanalytical measurements has increased in recent years because of their applicability to the determination of active compounds that undergo oxidation reactions,which is a matter of great importance in the field of clinical and pharmaceutical analysis.
4-Aminoantipyrine(AAP,as shown in Fig.1)is an aromatic substancewithanalgesic,antipyreticandanti-inflammatory properties[18].However,AAP usually produces side effects such as the risk of agranulocytosis[19].Although AAP is scarcely ever administered as an analgesic because of side effects,as a raw material,it is mostly used to produce 4-aminoantipyrine derivatives,which have better biological activities[20,21].In addition,it is used as a reagent for biochemical reactions producing peroxides or phenols[22,23]and can also be used to detect phenols in the environment[24].Since it is widely used in the pharmaceutical industry,biochemical research and environmental monitoring,AAP has become an environmental pollutant.
The toxic effect of AAP on experimental animals was reported[25].AAP can reduce blood flow[26]and 13,14-dihydro-15-keto prostaglandin F2 alpha concentration[27]after it is infused into the blood.AAP can form stable complexes with heme[28].
Different methods have been reported for the determination of AAP including liquid and gas chromatography,spectrophotometricmethod[29-31],liquid chromatography/mass spectrometry[32],capillary electrophoresis[33],solid phase spectrophotommetry[34],different HPLC methods[35-37]and voltammetric method by using graphite pencil electrode[38].The main problems encountered in using some methods are time-consuming extraction and separation procedures.
Carbon nanotubes have several applications in the field of semiconductor devices,high performance nano-composites,energy conversion devices,sensors,etc.[39,40]because of their nano-scale structure,large surface area,high mechanical strength and extraordinary electronic properties.There are so many results on the modification of the electrode surface using carbon nanotubes[41-44].
In this paper,we demonstrated a successful way to disperse multiwalledcarbonnanotube(MWCNT)withincetyltrimethylammonium bromide(CTAB)surfactant.In this procedure,the surfactant is adsorbed on the surface of MWCNTs,and subsequent ultrasonication of the solution,which takes several minutes,will cleave apart their aggregations and debundle nanotubes by steric or electrostatic repulsions resulted from the charge of surfactant hydrophilic groups[45-47].The resulted electrochemical sensors exhibited high sensitivity,rapid response,good reproducibility,low detection limit,renewal of the surface and freedom from other potentially interfering species.
2.Materials and methods
2.1.Apparatus
Electrochemical studies were carried out by CH Instruments(Electrochemical Analyzer,Model 630D,USA),a three electrode system consisting of a glassy carbon electrode(GCE)modified with MWCN/CTAB as a working electrode,saturated Ag/AgCl/KCl as a reference electrode and a platinum wire as the counter electrode. Electrode surface morphology study was carried out by SEM instrument model OXFORD instrument INCA PENTA FETX3 CARL ZEISS(Japan)and Nanosurf Easyscan 2 atomic force microscopy(AFM)(Switzerland).An Elico LI-120pH meter(Elico Ltd.Hyderabad,India)was used to determine the pH of the buffer solution.
2.2.Reagents and chemicals
4-Aminoantipyrine and MWCNT powders were purchased from Sigma-Aldrich(Mumbai,India).Cetyltrimethylammonium bromide was from Merck(Bengaluru,India).Double distilled water was used throughout the work.All other solvents and materials used throughout this study were of analytical grade.
2.3.Preparation of modified electrode
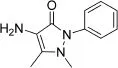
Fig.1.Chemical structure of 4-aminoantipyrine.

Fig.2.Schematic diagram of the proposed modification steps.
To get reproducible results,great care was taken in the electrode pre-treatment.The GCE was pre-treated in two ways:(i)mechanical polishing over a velvet micro-cloth with 0.3 and 0.05 μm alumina slurry and(ii)electrochemical treatment by applying a potential of 1.25 V for 10 s vs.Ag/AgCl.The electrochemical pre-treatment was done in the same supporting electrolyte solution in which the measurement was carried out.After that 10 μL of solution containing 0.3 g/L MWCNTs and 0.2 g/L CTAB,which was sonicated for 60 min,was placed on the GCE surface and then evaporated in an oven at 50°C.The ultrasonication of MWCNTs via CTAB will lead to the dispersion of nanotubes,and fix the surfactants on the surface of MWCNTs(possible arrangements of CTAB on MWCNTs are illustrated in Fig.2[48]).It can be described that the cationic surfactant will makethenanotubes positively charged,and thesecharged MWCNTs are driven toward cathode to form a thin layer at the electrode surface.
Eventually,the coated electrodes(MWCNTs-CTAB/GCE)were immersed in the bicarbonate solution(0.01 M)for 30 min in order to extract the residual surfactants from the surface of electrode. The modified electrodes were washed with distilled water and dried at room temperature(MWCNTs-GCE).Fig.3 shows surface morphology of photography of SEM images of unmodified and modified GCE,and AFM image of modifier.
The area of the electrode was calculated by the cyclic voltammetric method using 1.0 mM K3Fe(CN)6in 0.1 M KCl by recording the current voltage curve at different scan rates(Supplementary Fig.1).For a reversible process,the following Randles-Sevcik formula can be used[49].
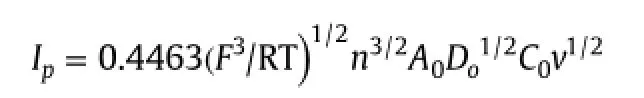
where Iprefers to the cathodic peak current,n is the number of electrons transferred,A0is the surface area of the electrode,D0is diffusion coefficient,v is the scan rate and C0is the concentration of K3Fe(CN)6.For 1.0 mM K3Fe(CN)6in 0.1 M KCl electrolyte,n=1,DR=7.6×10-6cm2/s,then from the slope of the plot of Ipversus v1/2,the electro active area was calculated.In our experiment,the slope was 0.695 and the area of electrode was calculated to be 0.117 cm2.The area of the unmodified glassy carbon electrode was calculated to be 0.0448 cm2.
2.4.Plasma sample preparation
Human plasma sample was prepared as described in the earlier report of our work[50].Human blood samples were collected in dry and evacuated tubes(which contained saline and sodium citrate solution)from a healthy volunteer.The samples were handled at room temperature and centrifuged for 10 min at 1500 rpm for the separation of plasma within 1 h of collection.The samples were then transferred to polypropylene tubes and stored at -20°C until analysis.The plasma samples,0.2 mL,were deprotonised with 2 mL of methanol.After vortexing for 15 min,themixture was then centrifuged for 15 min at 6000 rpm,and supernatants were collected.
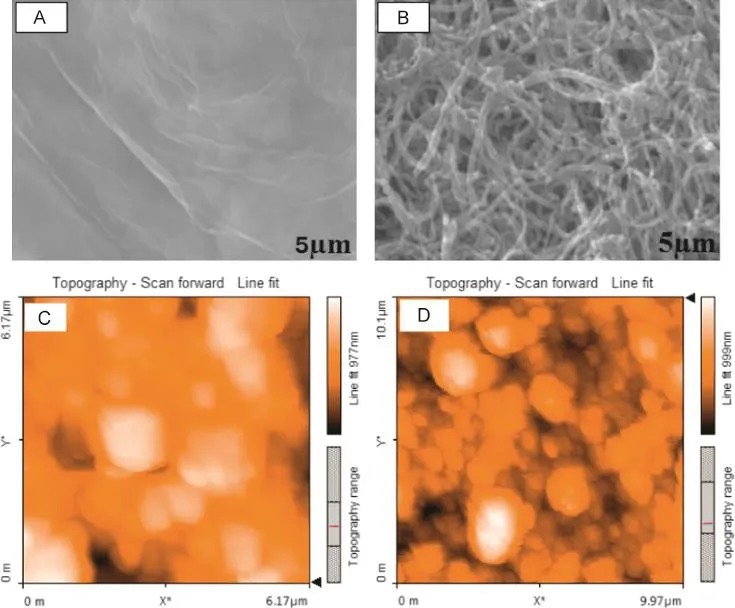
Fig.3.SEM images of bare glassy carbon electrode and MWCN-CTAB modified glassy carbon electrode.(A)SEM image of bare GCE,(B)SEM image of MWCNT-CTAB modified GCE,(C)AFM topography of MWCNTs,and(D)AFM topography of MWCNT-CTAB sample.
2.5.Pharmacokinetic study
Serum samples of a healthy volunteer were collected as described in Section 2.4 and stored at-20°C until analysis.Into each of 10 centrifugation tubes(3 mL polypropylene microcentrifuge tubes)containing 1.0×10-8M concentration of AAP,100 μL volume of the human serum was transferred,then mixed well with 1 mL of methanol to denature and precipitate proteins.The solutions were centrifuged for 3 min at 14,000 rpm to separate out the precipitated proteins.The clear supernatant layers of these solutions were filtered through 0.45 μm millipore filters to produce protein-free human serum samples.Each sample was analyzed at different time intervals by using differential pulse voltammetry.
3.Results and discussion
3.1.Cyclic voltammetric study of 4-aminoantipyrine
The electrochemical response of 0.1 mM AAP was investigated by cycle voltammetry between 0.2 and 0.8 V in phosphate buffer solution of pH 3.0 at GCE,MWCNT/GCE and MWCNT-CTAB/GCE(Fig.4).At GCE,a poorly defined oxidation peak was observed and the peak current was smaller.AAP exhibited well defined anodic peak at 0.512 V at MWCNT/GCE.The oxidation peak current increased greatly at MWCNT-CTAB/GCE(the voltammogram as shown in Fig.4).It indicates that MWCNT-CTAB/GCE can make the electron transfer of AAP easily.No reduction peak was observed in the reverse scan,suggesting that the electrochemical reaction was totally irreversible process.
3.2.Effect of amount of MWCNT-CTAB suspension
We examined the effect of MWCNT-CTAB suspension amount on the electrochemical behavior of AAP.The results suggested that the amount of MWCNT-CTAB suspension influenced the current responses of AAP.Supplementary Fig.2 demonstrates the relationship between the oxidation peak current of AAP and the amount of MWCNT-CTAB suspension used for coating GC electrode.As can be seen,the peak current gradually increased with increasing the amount of MWCNT-CTAB suspension from 0 to 10 μL,owing to the increased effective electrode surface area for AAP oxidation.Further increasing the amount of MWCNT-CTAB suspension,the peak current almost remained stable.However,when it exceeded 14 μL,the peak current slightly decreased.When the coating film was too thick,the film no longer adhered tightly to the glass carbon,reducing conductivity and part of the MWCNTCTAB left the electrode surface.More excessively coated amount of MWCNT-CTAB suspension led to less adherent film.Accordingly,10 μL of MWCNT-CTAB suspension solution providing the maximum current response was used in further experiments,while the amount of suspension of MWCNT-CTAB had little effect on the oxidation potential of AAP.
3.3.Effect of pH
The pH of the supporting electrolyte had a noticeable effect on the electro-oxidation of analyte under investigation.The electrooxidation of AAP was carried out by cyclic voltammetry at the surface of MWCNT-CTAB modified GCE over the pH range 3.0-7.0. The peak potential shifted to more negative values with an increase in solution pH(Fig.5).The sharp and well-defined oxidation peak was observed in phosphate buffer of pH 3.Hence,we selected phosphate buffer of pH 3 for further studies.
3.4.Effect of scan rate
The cyclic voltammograms of 0.1 mM AAP on the MWCNTCTAB modified GCE at different scan rates are shown in Fig.6A. The observation was made to investigate the kinetics of the electrode reaction.With the increase of the scan rate,the oxidation peak current also increased gradually,indicating the direct electron transfer between AAP and modified electrode surface.In the range from 10 to 100 mV/s the oxidation peak current was proportional to the scan rate(Ipa(10 μA)=0.043-0.099 v1/2)and the correlation coefficient was 0.984,which indicated that the electron transfer reaction was a diffusion-controlled process[51].A linear relationship was observed between log Ipaand log v(log Ipa(μA)= 0.3088 log v(V/s)+1.275;r=0.976).The slope value of 0.3088 confirmsthattheelectro-oxidationofAAPwasdiffusion controlled.
A linear relation between peak potential(Epa)and log ν was obtained,Epa=0.018 log v+0.445(Fig.6B).Such behavior revealed the irreversible nature of the electrochemical process for AAP. According to Laviron,for a diffusion-controlled irreversible process[52],Epais defined by the following equation:
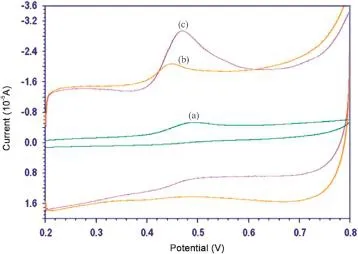
Fig.4.Typical cyclic voltammograms of 0.1 mM 4-aminoantipyrine at(a)bare GCE,(b)GCE+MWCNT and(c)GCE+MWCNT-CTAB.
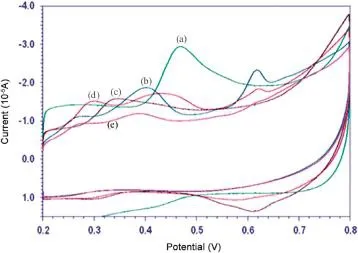
Fig.5.Influence of pH on the electro-oxidation of 0.1 mM of AAP.(a)-(e):3.0,4.2,5.0,6.2,7.0.
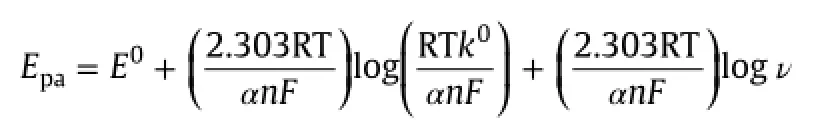
where α is the transfer coefficient,k0is the heterogeneous electron transfer rate constant of the reaction,n is the number of transferred electrons,v is the potential scan rate and E0is the formal redox potential.The other symbols have their usual meaning.This relationship allows n to be readily calculated from the slope of the Epavs.logvplot.TakingT=298 K,R=8.314 J/Kmoland F=96480 C and the value α was calculated from Bard and Fualkner equation,which is equal to 1.325,the value of n was calculated to be 2.47≈2 for AAP.
If E0is known,the value of k0can be estimated from the intercept of the above plot.E0in the above equation can be obtained from the ordinate intercept of the Epavs.v curve at v=0[53].The obtained k0was 9.845×103/s.
3.5.Calibration curve
In order to develop the voltammetric method for determination of the drug,we chose the differential pulse voltammetric method for the reason that the peaks were sharper and more distinct at a lower concentration of AAP than those obtained by CV,with a lower background current,resulting in enhanced resolution.In keeping with the obtained results,it was feasible to apply this technique to the quantitative analysis of AAP.The phosphate buffer solution of pH 3.0 was selected as the supporting electrolyte for the quantification as AAP gave maximum peak current.Differential pulse voltammograms obtained with increasing amounts of AAP showed that the peak current increased linearly with increased concentration.Linear calibration curves were obtained for AAP concentration in the range of 5.0×10-9-4.0×10-8M(Fig.7).Linear equation was Ipa=4.47(10-8M)-0.606(r=0.988).Interrelated statistical data of the calibration curves were procured from the three different calibration curves.Limits of detection(LOD)and quantification(LOQ)were calculated based on the peak current using the following equations: where s is the standard deviation of the peak currents of the blank(three replicates),and m is the slope of the calibration curve.The LOD and LOQ values were 1.63×10-10M,and 5.42×10-10M,respectively.The detection limits reported at different analytical methods for AAP related dipyrone derivative drugs are tabulated in Table 1.The proposed method was better than other reported electrochemical methods[31,38,54-56].

3.6.Robustness and effect of excipients
The robustness of the method was checked by evaluating the influence of small variations of some of the most important variables,including pH,accumulation time and potential range.The results indicated that none of these variables significantly affected the recovery of AAP.This provided an indication of the dependability of the proposed process for the assay of AAP,and the proposed method could be measured robustly.
For the possible analytical application of the proposed method,the effect of some common excipients used in pharmaceutical preparations was examined.The tolerance limit was defined as the maximum concentration of the interfering substance that caused an error less than 5%for determination of AAP.The experimental results showed that hundred-fold excess of citric acid,dextrose,glucose,gum acacia,lactose,tartaric acid and sucrose did not interfere with the voltammetric signal of AAP.This showed that the electrode was much selective towards AAP.
3.7.Reproducibility of the modified electrode
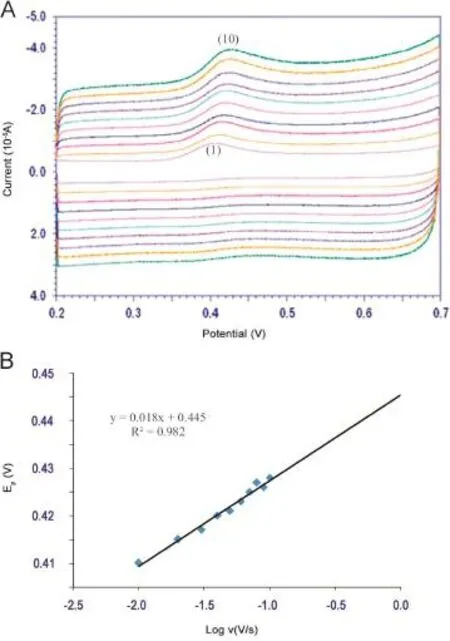
Fig.6.(A)Cyclic voltammograms of 0.1 mM AAP at different scan rates(1-10:10,20,30,40,50,60,70,80,90,100 mV/s)in 0.2 M phosphate buffer(pH 3.0).(B)Relationship between peak potential and logarithm of scan rate.
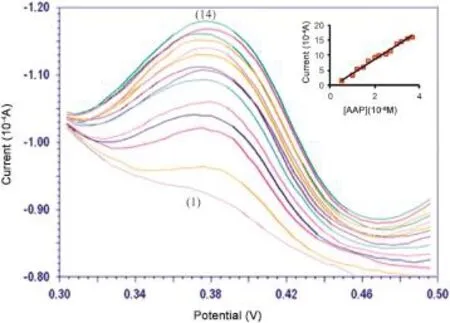
Fig.7.Differential pulse voltammograms of AAP at different concentrations at MWCNT-CTAB/GCE:(1)-(14)0.5×10-8-4.0×10-8M.Inset:plot of the concentration vs.peak current of AAP at MWCNT-CTAB/GCE.
The renewal and reproducibility of the electrode were investigated.It was found that after determination the surface of the MWCNT-CTAB/GCE could be regenerated by successively cycling between 0 and 1.2 V in 3.0 pH with 0.2 M phosphate buffer afterwashing the electrode with water and acetone.As an example,0.1 mM AAP solution was measured successively for 5 times with the same electrode regenerated through such procedure after every determination,the relative standard deviation(RSD)of the peak current was 1.15%.As to the reproducibility between some days,it was similar to that of within a day,if the temperature was kept almost unchanged.Peak current obtained for 0.1 mM AAP solution on different days is shown in Supplementary Fig.3.Owing to the adsorption of AAP or its reductive products on to the electrode surface,the current response of the modified electrode would decrease after successive use.In this case,the electrode should be prepared again.
3.8.Determination of AAP in biological samples
The applicability of the proposed method for the determination of AAP in biological fluid of human urine and plasma samples was attempted.Drug-free human urine and plasma samples,obtained from healthy volunteers,were filtrated through a filter paper and stored frozen until the assay.The developed differential pulse voltammetric method for the AAP determination was applied to urine and plasma samples.The recoveries from urine and plasma were measured by spiking drug-free urine and plasma with known amounts of AAP.The urine and plasma samples were diluted with the phosphate buffer solution before analysis without further pretreatment.A quantitative analysis could be carried out by adding the standard solution of AAP into the detection system of urine and plasma samples.The calibration graph was used for the determination of spiked AAP in urine samples.The detection results of urine and plasma samples are listed in Table 2.The recovery determined was in the range of 98.02%-103.46%for human plasma and 99.16%-102.17%for urine sample.Thus,satisfactory recoveries of the analytes from the real samples and a good agreement between the concentration ranges studied and the real ranges encountered in the urine and plasma samples when treated with the drug made the developed method applicable in clinical analysis.
浅谈加强企业青年人才队伍建设……………………………………………………………………………………李成丽(4.88)
3.9.Application to pharmacokinetic studies
Pharmacokinetics is the study of the time course of drug absorption,distribution,metabolism,and excretion.Clinical pharmacokinetics is the application of pharmacokinetic principles to the safe and effective therapeutic management of drugs in an individual patient.Primary goals of clinical pharmacokinetics include enhancing efficacy and decreasing toxicity of a patient's drug therapy.The development of strong correlations between drug concentrations and their pharmacologic responses has enabled clinicians to apply pharmacokinetic principles to actual patient situations.A drug's effect is often related to its concentration at the site of action,so it would be useful to monitor this concentration.Receptor sites of drugs are generally inaccessible to our observations or are widely distributed in the body,and therefore direct measurement of drug concentrations at these sites is not practicable.However,drug concentration in the blood or plasma,urine,saliva and other easily sampled fluids can be measured.
The assay results are shown in Supplementary Fig.4,which illustrates the profile of the plasma concentration vs.time for AAP. The results suggested that the disposition of AAP was conformable to a one compartment open model.Table 3 shows the peak response of drug concentration at different time intervals and some the pharmacokinetic parameters for AAP in the plasma sample.

Table 1Comparison of detection limits for AAP related dipyrone derivative drugs by different methods.
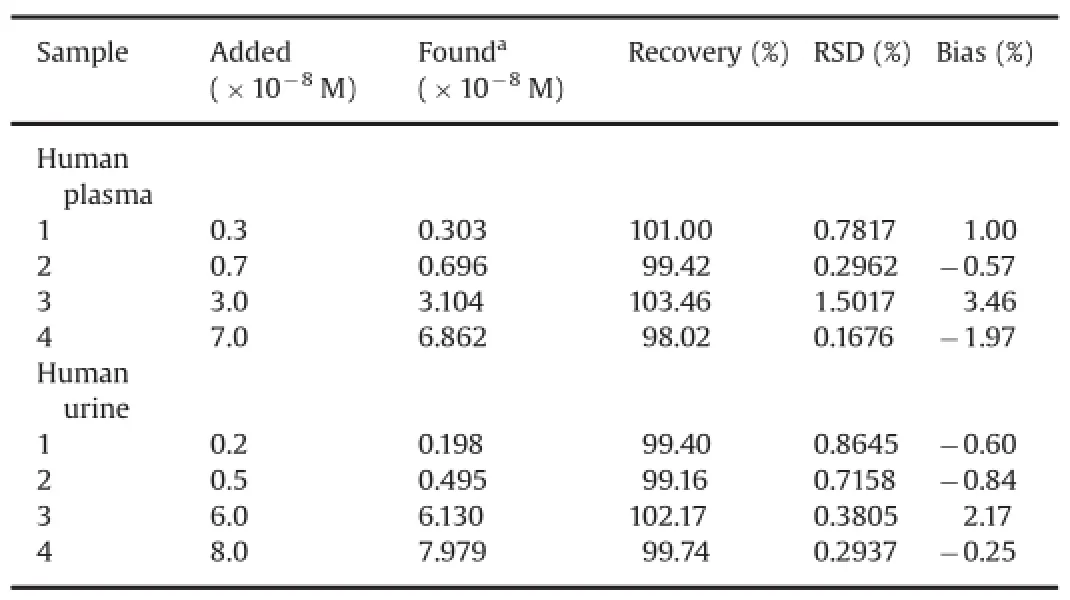
Table 2Application of DPV to the determination of AAP in spiked human urine and blood plasma samples.

Table 3Response of peak current of 1×10-8M AAP in urine sample at different time intervals.
4.Conclusion
The voltammetric behavior and oxidation mechanism of AAP were investigated at an MWCN-CTAB/GCE by CV in phosphate buffer solution of pH 3.0.Based on this study,influences of several physicochemical parameters such as potential scan rate,pH and concentration were investigated.The oxidation of AAP was found to be an irreversible with diffusion character.MWCNT-CTAB/GCE showed electrocatalytic action for the oxidation of AAP,characterizing by the enhancement of the peak current,which was probably due to the larger effective surface area of MWCNT-CTAB. This method was successfully used to determine AAP in the human urine and plasma samples.The proposed method offered the advantages of accuracy and time saving as well as simplicity of reagents and apparatus.In addition,the results obtained in the analysis of AAP in spiked urine and plasma samples demonstrated the applicability of the method for real sample analysis.
Acknowledgments
One of the author(J.I.Gowda)thanks UGC,New Delhi,India,for the award of Research Fellowship in Science for Meritorious Students(RFSMS).
Appendix A.Supplementary Information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.jpha.2015.01.001.
References
[1]B.Nigovic,B.Simunic,Determination of 5-aminosalicylic acid in pharmaceutical formulation by differential pulse voltammetry,J.Pharm.Biomed.Anal.31(2003)169-174.
[3]A.K.Jain,V.K.Gupta,S.Radi,et al.,A comparative study of Pb2+sensors based on derivatized tetrapyrazole and calix[4]arene receptors,Electrochim.Acta 51(12)(2006)2547-2553.
[4]A.K.Jain,V.K.Gupta,L.P.Singh,et al.,Macrocycle based membrane sensors for the determination of cobalt(II)ions,Analyst 122(1997)583-586.
[5]J.Wang,Electroanalytical Techniques in Clinical Chemistry and Laboratory Medicine,VCH,New York,1988.
[6]V.K.Gupta,B.Sethi,R.A.Sharma,et al.,Mercury selective potentiometric sensor based on low rim functionalized thiacalix[4]-arene as a cationic receptor,J.Mol.Liq.177(2013)114-118.
[7]R.N.Goyal,V.K.Gupta,S.Chatterjee,Electrochemical oxidation of 2′,3′-dideoxyadenosine at pyrolytic graphite electrode,Electrochim.Acta 53(2008)5354-5360.
[8]V.K.Gupta,A.K.Jain,G.Maheshwari,et al.,Copper(II)-selective potentiometric sensors based on porphyrins in PVC matrix,Sens.Actuators B 117(2006)99-106.
[9]Y.Tang,C.Sun,X.Yang,et al.,Graphene modified glassy carbon electrode for determination of trace aluminium(III)in biological samples,Int.J.Electrochem.Sci.8(2013)4194-4205.
[10]B.P.Bator,L.Cabaj,M.Raś,et al.,Potentiometric sensor platform based on a carbon black modified electrodes,Int.J.Electrochem.Sci.9(2014)2816-2823.
[11]R.N.Goyal,V.K.Gupta,N.Bachheti,Fullerene-C60-modified electrode as a sensitive voltammetric sensor for detection of nandrolone—an anabolic steroid used in doping,Anal.Chim.Acta 597(2007)82-89.
[12]B.H.Chiou,Y.T.Tsai,C.M.Wang,Phenothiazine-modified electrodes:a useful platform for protein adsorption study,Langmuir 30(2014)1550-1556.
[13]Q.M.Feng,Q.Zhang,C.G.Shi,et al.,Using nanostructured conductive carbon tape modified with bismuth as the disposable working electrode for stripping analysis in paper-based analytical devices,Talanta 115(2013)235-240.
[14]S.H.Yu,G.C.Zhao,Preparation of platinum nanoparticles-graphene modified electrode and selective determination of rutin,Int.J.Electrochem.2012(2012)431253-431259.
[15]Y.Miao,J.Chen,X.Wu,Electrochemical behaviors of matrine at l-cysteinemodified electrodes,Surf.Rev.Lett.115(2008)537-543.
[16]A.Balamurugan,S.M.Chen,Silver nanograins incorporated pedot modified electrode for electrocatalytic sensing of hydrogen peroxid,Electroanalysis 21(2009)1419-1423.
[17]R.N.Goyal,V.K.Gupta,N.Bachheti,et al.,Electrochemical sensor for the determination of dopamine in presence of high concentration of ascorbic acid using a fullerene-C60coated gold electrode,Electroanalysis 20(2008)757-764.
[18]Y.M.Chen,Y.P.Chen,Measurements for the solid solubilities of antipyrine,4-aminoantipyrine and 4-dimethylaminoantipyrine in supercritical carbon dioxide,Fluid Phase Equilib.282(2009)82-87.
[19]A.Lang,C.Hatscher,C.Wiegert,et al.,Protease-catalysed coupling of Nprotected amino acids and peptides with 4-aminoantipyrine,Amino Acids 36(2009)333-340.
[20]S.Cunha,S.M.Oliveira,M.T.Rodrigues,et al.,Structural studies of 4-aminoantipyrine derivatives,J.Mol.Struct.752(2005)32-39.
[21]S.Prasad,R.K.Agarwal,Cobalt(II)complexes of various thiosemicarbazones of 4-aminoantipyrine:syntheses,spectral,thermal and antimicrobial studies,Transit.Met.Chem.32(2007)143-149.
[22]J.F.Van Staden,N.W.Beyene,R.I.Stefan,et al.,Sequential injection spectrophotometric determination of ritodrine hydrochloride using 4-aminoantipyrine,Talanta 68(2005)401-405.
[23]J.Kasthuri,J.Santhanalakshmi,N.Rajendiran,Platinum nanoparticle catalysed coupling of phenol derivatives with 4-aminoantipyrine in aqueous medium,Transit.Met.Chem.33(2008)899-905.
[24]C.Z.Katsaounos,E.K.Paleologos,D.L.Giokas,et al.,The 4-aminoantipyrine method revisited:determination of trace phenols by micellar assisted preconcentration,Int.J.Environ.Anal.Chem.83(2003)507-514.
[25]A.M.Vinagre,E.F.Collares,Effect of 4-aminoantipyrine on gastric compliance and liquid emptying in rats,Braz.J.Med.Biol.Res.40(2007)903-909.
[26]S.G.Sunderji,A.El Badry,E.R.Poore,et al.,The effect of myometrial contractures on uterine blood flow in the pregnant sheep at 114 to 140 days' gestation measured by the 4-aminoantipyrine equilibrium diffusion technique,Am.J.Obstet.Gynecol.149(1984)408-412.
[27]A.El Badry,J.P.Figueroa,E.R.Poore,et al.,Effect of fetal intravascular 4-aminoantipyrine infusions on myometrial activity(contractures)at 125 to 143 days'gestation in the pregnant sheep,Am.J.Obstet.Gynecol.150(1984)474-479.
[28]S.C.Pierre,R.Schmidt,C.Brenneis,et al.,Inhibition of cyclooxygenases by dipyrone,Br.J.Pharmacol.151(2007)494-503.
[29]E.Emerson,Standard Methods for the Examination of Water and Waste Wáter,17th ed.,American Public Health Association,New York,1989,pp.5-51.
[30]P.Majlat,Gas chromatography determination of atropine,theophylline,phenobarbital and aminophenazone in tablets,Pharmazie 39(1984)325-326.
[31]L.Penney,C.Bergeron,B.Coates,et al.,Simultaneous determination of residues of dipyrone and its major metabolites in milk,bovine muscle,and porcine muscle by liquid chromatography/mass spectrometry,J.AOAC Int.88(2005)496-504.
[32]D.Puig,I.silgoner,M.Grasserbauer,et al.,Part-per-trillion level determination of priority methyl-,nitro-,and chlorophenols in river water samples by automated on-line liquid/solid extraction followed by liquid chromatography/ mass spectrometry using atmospheric pressure chemical ionization and ion spray interfaces,Anal.Chem.69(1997)2756-2761.
[33]E.Dabek-Zlotorzynska,Capillary electrophoresis in the determination of pollutants,Electrophoresis 18(1997)2453-2464.
[34]N.Isoshi,N.Sachico,W.Kaori,et al.,Determination of phenol in tap water and river water samples by solid phase spectroscopy,Anal.Sci.16(2000)269-274.[35]G.Blo,F.Dondi,A.Betti,et al.,Determination of phenols in water samples as 4-aminoantipyrine derivatives by high-performance liquid chromatography,J. Chromatogr.A 257(1983)69-79.
[36]D.Damm,Simultaneous determination of the main metabolites of dipyrone by high-pressure liquid chromatography,Arzneimittelforschung 39(1989)1415-1417.
[37]I.Carretero,J.M.Vadillo,J.J.Laserna,Determination of antipyrine metabolites in human plasma by solid-phase extraction and micellar liquid chromatography,Analyst 120(1995)1729-1732.
[38]J.I.Gowda,S.T.Nandibewoor,Electrochemical behavior of 4-aminophenazone drug at graphite pencil electrode and its application in real samples,Ind.Eng. Chem.Res.51(2012)15936-15941.
[39]H.Zhou,T.Wang,Y.Y.Duan,A simple method for amino-functionalization ofcarbon nanotubes and electrodeposition to modify neural microelectrodes,J. Electroanal.Chem.688(2013)69-75.
[40]F.Fathirad,D.Afzali,A.Mostafavi,et al.,Fabrication of anew carbon paste electrode modified with multi-walled carbon nanotube forstripping voltammetric determination of bismuth(III),Electrochim.Acta 103(2013)206-210.
[41]B.Dogan-Topal,B.Bozal-Palabıyık,B.Uslu,et al.,Multi-walled carbonnanotube modified glassy carbon electrode as a voltammetric nanosensor forthe sensitive determination of anti-viral drug valganciclovir in pharmaceuticals,Sens.Actuators B 177(2013)841-847.
[42]S.Shahrokhian,M.Ghalkhani,M.Adeli,et al.,Multi-walled carbon nano-tubes with immobilised cobalt nanoparticle for modification of glassy carbonelectrode:application to sensitive voltammetric determination of thioridazine,Biosens.Bioelectron.24(2009)3235-3241.
[43]E.Baldrich,R.Gómez,G.Gabriel,et al.,Magnetic entrapment for fast,simple and reversible electrode modification with carbon nanotubes:applicationto dopamine detection,Biosens.Bioelectron.26(2011)1876-1882.
[44]R.N.Goyal,V.K.Gupta,S.Chatterjee,Simultaneous determination of adenosine and inosine using single-wall carbon nanotubes modified pyrolytic graphite electrode,Talanta 76(2008)662-668.
[45]L.Vaisman,H.D.Wagner,G.Marom,The role of surfactants in dispersion ofcarbon nanotubes,Adv.Colloid Interface Sci.37(2006)128-130.
[46]S.Swarup,C.K.Schoff,A survey of surfactants in coatings technology,Prog. Org.Coat.23(1993)1-22.
[47]M.Y.Pletnev,Chemistry of surfactants,in:V.B.Fainerman,D.Möbius,R.Miller(Eds.),Studies in Interface Science,vol.13,Elsevier,Amsterdam,2001,p.1.
[48]E.Pajootan,M.Arami,Structural and electrochemical characterization of carbon electrodemodified by multi-walled carbon nanotubes and surfactant,Electrochim.Acta 112(2013)505-514.
[49]D.I.Anguiano,M.G.Garcia,C.Ruíz,et al.,Electrochemical detection of iron in a lixiviant solution of polluted soil using a modified glassy carbon electrode,Int. J.Electrochem.2012(2011)1155-1160.
[50]J.C.Abbar,S.T.Nandibewoor,Development of electrochemical method for the determination of chlorzoxazone drug and its analytical applications to pharmaceutical dosage form and human biological fluids,Ind.Eng.Chem.Res.51(2012)111-118.
[51]R.N.Hegde,R.R.Hosamani,S.T.Nandibewoor,Electrochemical oxidation and determination of theophylline at a carbon paste electrode using cetyltrimethyl ammonium bromide as enhancing agent,Anal.Lett.42(2009)2665-2682.
[52]E.Laviron,General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems,J.Electroanal.Chem.101(1979)19-28.
[53]Y.Wu,X.Ji,S.Hu,Studies on electrochemical oxidation of azithromycin and its interaction with bovine serum albumin,Bioelectrochemistry 64(2004)91-97.
[54]R.L.C.Thiago Paixao,R.C.Matos,M.Bertotti,Diffusion layer titration of dipyrone in pharmaceuticals at a dual-band electrochemical cell,Talanta 61(2003)725-732.
[55]G.Gopalakrishnan,P.Manisankar,B.Muralidharan,et al.,Stripping voltammetric determination of analgesics in their pharmaceuticals using nano-riboflavin-modified glassy carbon electrode,Int.J.Electrochem.2011(2011)1-11.
[56]L.R.Cumba,U.O.Bicalho,D.R.Silvestrini,et al.,Preparation and voltammetric study of a composite titanium phosphate/nickel hexacyanoferrate and its application in dipyrone determination,Int.J.Chem.4(2012)66-78.
11 September 2014
in revised form
☆Peer review under responsibility of Xi'an Jiaotong University.
.Tel.:+91 836 2770524;fax:+91 836 2747884.
E-mail address:stnandibewoor@yahoo.com(S.T.Nandibewoor).
http://dx.doi.org/10.1016/j.jpha.2015.01.001
2095-1779/©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Quantitative bioanalytical and analytical method development of dibenzazepine derivative,carbamazepine:A review☆
- Development and validation of a GC-FID method for quantitative analysis of oleic acid and related fatty acids☆
- Simultaneous determination of four Sudan dyes in rat blood by UFLC-MS/MS and its application to a pharmacokinetic study in rats☆
- Multi-spectroscopic investigation of the binding interaction of fosfomycin with bovine serum albumin☆
- Identification,synthesis and characterization of process related impurities of benidipine hydrochloride,stress-testing/stability studies and HPLC/UPLC method validations☆
- Simultaneous quantitation of folic acid and 5-methyltetrahydrofolic acid in human plasma by HPLC-MS/MS and its application to a pharmacokinetic study☆