3个VHL综合征家族的基因检测及临床调查
2015-03-02吴鑫尧陈江明赵义军刘付宝耿小平
吴鑫尧,陈江明,赵义军,谢 坤,刘付宝,耿小平
3个VHL综合征家族的基因检测及临床调查
吴鑫尧1,陈江明2,赵义军1,谢 坤1,刘付宝1,耿小平1
摘要目的 明确von Hippel-Lindau综合征(VHL综合征)家族基因突变及临床表现特点,筛查出家族内基因突变携带者行临床筛查,综合家族发病特点行家族健康指导。方法
通过流行病学调查,共3例先证者临床诊断VHL综合征。抽取先证者及家族自愿者外周血,应用聚合酶链反应(PCR)体外扩增得到vhl基因片段,通过测序得到基因信息;再对家族内具有vhl基因突变的携带者进行头颅MRI、腹部B超等对全身多系统行临床筛查;综合家族发病特点给出家族健康指导。结果 3例先证者均发现vhl基因发生突变,家族1 中5例成员基因阳性,突变方式为外显子1 c.330C>A;家族2先证者外显子3488delC;家族3发现7例基因阳性者,突变方式为外显子1 c.233G>A。其中,家族成员1III3、3III1、3III4、3IV3确定为基因突变携带者,临床检查显示其中3例成员已发病,1III3表现为双肾多发囊肿,胰腺多发囊肿;3III1表现为胰腺多发囊肿;3III4表现为视网膜血管母细胞瘤、胰腺多发囊肿,视网膜血管母细胞瘤接受激光治疗。3IV3由于年龄较小,临床检查未见明显异常。所有基因携带者接受规范化随访。结论 基因检测可早期确诊VHL综合征,临床上对vhl基因突变患者需进行严密随访,从而提高患者治疗效果、延长生存期或改善生活质量。
关键词VHL综合征;vhl基因;基因检测
2015-02-13接收
作者单位:1安徽医科大学第一附属医院器官移植中心,合肥230022
2安徽医科大学第二附属医院普外科,合肥 230601
von Hippel-Lindau综合征(VHL综合征)是一种常染色体显性基因遗传性疾病,是染色体3P25 (OMIM 193300)长约10 bp的VHL肿瘤抑制基因突变所致。发生率约1/36 000,60岁之前,基因外显率达95%[1]。临床上常表现为视网膜及中枢神经系统的血管母细胞瘤、肾透明细胞癌、嗜铬细胞瘤和胰腺肿瘤。尽管目前对该病分子生物学的认识以及手术时机的选择有所进展,但VHL综合征患者的平均生存年龄仅49岁[2]。该研究通过对3例临床诊断VHL综合征的患者及其家族内27例自愿者行vhl基因检测,报道如下。
1 材料与方法
1.1 病例资料 安徽医科大学第一附属医院2011 年1月~2013年12月有3例患者临床诊断为VHL综合征,3个家族共31例成员均自愿加入本项研究,签署知情同意书,抽取每个家族成员外周血5 ml,于-80℃冰箱保存,并收集临床资料。
1.2 实验方法
1.2.1 DNA提取 用血液基因组DNA提取试剂盒Flexigene DNA kit(Qiagen,51204)提取外周血DNA,并以电泳检测提取DNA纯度,检测条件为150 V、100 mA电脉20 min,见图1。
1.2.2 聚合酶链式反应(polymerase chain reactions,PCR) 用Primer 5.0软件设计引物,引物涵盖VHL基因全部3个外显子编码区域,引物由上海生工生物工程技术有限公司合成,见表1。PCR将基因组DNA(100~200 ng)扩增,25 μl PCR反应体系包括:15.5 μl蒸馏水,5 μl 5×buffer缓冲液,1 μl脱氧核苷三磷酸(5 mmol/L),1 μl DNA模板,0.5 μl DNA聚合酶,40 mmol/L的上下游引物各1 μl。PCR反应扩增条件:外显子1 95℃预变性10 min,95℃30 s,62℃30 s,72℃20 s,共35个循环,最终72℃下延伸10 min。外显子2和外显子3在60℃下行PCR扩增。1%琼脂糖电泳,150 V、100 mA电泳20 min后使用紫外凝胶成像仪进行PCR结果检测。
1.2.3 PCR反应纯化及测序 PCR产物经AxyPrep-96 Kit纯化后送生工测序。
1.2.4 测序结果分析 NCBI数据库(http://www.ncbi.nlm.nih.gov/)中找到VHL基因,其包含3个外显子共642个密码子,编码214个氨基酸(包含末端不翻译氨基酸的3个终止密码子),Multalin方法对测得DNA序列比对(http://multalin.toulouse.inra.fr/multalin/)。测得序列在http://web.expasy.org/translate/中将可能表达氨基酸翻译,并与NCBI数据库中进行比对。

表1 vhl基因外显子片段大小和用于PCR扩增及测序的引物信息
1.3 临床检查方法 行VHL家族调查,并对家族内基因突变携带者行头颅MRI、B超(肝、胆、胰、脾、肾、肾上腺及盆腔)、眼底、血尿常规及尿儿茶酚胺检测。
2 结果
2.1 基因检测结果 PCR产物琼脂糖凝胶电泳结果显示均获得预期大小的扩增片段。3个家族31例健在成员均自愿接受基因分析,并绘制树状图。结果包括先证者在内,家族1中5例成员vhl基因阳性,家族2中1例,家族3中7例。4例(1Ⅲ3、3Ⅲ1、3Ⅲ4、3Ⅳ3)家族成员确定为基因突变携带者,3个家族基因突变方式分别为:家族1外显子1第330位碱基A替换为碱基C,导致vhl基因编码的第110个氨基酸组胺酸变为脯氨酸。家族2外显子3 第488位碱基C缺失,结果致vhl编码的氨基酸162位以后全部紊乱。家族3外显子1第233位碱基A转换为碱基G,导致vhl基因编码的第98个氨基酸天冬酰胺酸变为丝氨酸,见表2。
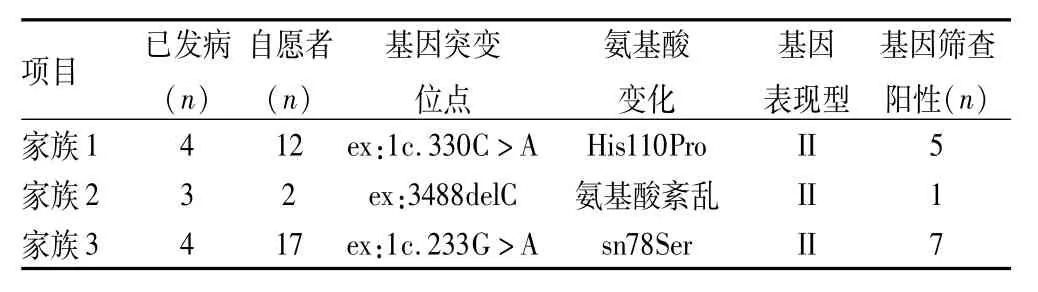
表2 3个家族基因突变位点、家系基因筛查结果以及VHL综合征发病类型
2.2 临床检查
2.2.1 家族调查 家族1:先证者1Ⅱ1,男,48岁,
1991年因进行性的头痛入安徽医科大学第一附属医院行“小脑血管母细胞瘤切除”,术后恢复良好,至今无复发。患者2003年出现肉眼血尿,行“左肾透明细胞癌剜除术”,术后入安徽医科大学第一附属医院泌尿外科二病区规范随访,2007年行“右肾透明细胞癌剜除术”,2012年行“左腰部转移瘤切除+左侧肾脏肿瘤射频消融”,术后不久发现左侧腰部肿块复合,呈进行性增大,考虑肾透明细胞癌转移,2014年1月行“左腰部肿瘤活检+左肾肿瘤射频消融术”,术后镜检于腰部脂肪组织见透明细胞肿瘤结节。患者1I1因多年头痛,48岁去世。患者1 II3,47岁,10年前因头晕发现肾上腺嗜铬细胞瘤,双肾多发囊肿,一直保守治疗。2012年因双肾囊肿于南京军区总医院行双肾囊肿开窗引流术,治疗效果不佳,囊肿无法完全根治,在患者诊治期间,其女儿(1III6),23岁,医生建议后体检发现多囊肝,双肾多发囊肿,至今尚无明显临床症状。先证者儿子(1III1),25岁,2年前因头晕,行走不稳,入安徽医科大学第一附属医院诊断小脑血管母细胞瘤,手术切除肿瘤,至今无复发。
家族2:先证者2II2,男,45岁,2013年12月因出现肉眼血尿半月余,安徽医科大学第一附属医院诊断多发肾脏占位、肺部转移可能、双侧肾上腺占位,失去治疗时机,现肿瘤前列腺、骨多发转移,于合肥市第五人民医院保守对症治疗,一般状况极差,见图3A。患者2 I2,23岁体检发现肾上腺占位、多发胰腺囊肿、多囊肾,并于上海市中山医院行肾上腺嗜铬细胞瘤切除术,胰腺囊肿未予处理。49岁出现血尿,诊断肾癌多发转移,50岁去世。患者2II1,为先证者孪生哥哥,2011年因头晕,行头颅MRI见左侧小脑半球囊性病变,于北京天坛医院行小脑肿瘤切除术,术后证明为血管母细胞瘤。2013年因肾透明细胞癌多发转移去世。
家族3:先证者3III2,女,27岁,2007年因行走不稳诊断小脑肿瘤,见图3B。手术切除后2010年、2013年两次复发,再次手术治疗。先证者左眼视物不清7年,失明1年,一直未予诊治,近期出现左眼胀痛,发作频繁,经指导后检查发现左眼眼底反光异常,正常红光反射消失,网膜可见不规则渗出病灶,静脉淤曲明显,并见自视乳头颞侧生成粗大扭曲滋养血管,并见后极部增殖膜,远端见瘤体,见图3C。先证者父辈发病3例,患者3II1 58岁,2014年2月因肉眼血尿1 d于淮南市人民医院就诊,诊断双肾多发肾癌晚期,失去治疗时机,纳入随访。患者3II2,1990年因头晕行小脑肿瘤切除术,1993年肿瘤复发,再次手术切除,因术后出血死亡。患者3II5,15年前因头痛、头晕急性发作,入淮南市矿三院,诊断双肾上腺嗜铬细胞瘤、小脑肿瘤,治疗期间颅内出血死亡。家族成员3III1平日偶感腹胀,症状可自行缓解,未予诊治。
2.2.2 临床检查 对4例(1III3、3III1、3III4、3IV3)基因突变携带者进行头颅MRI(3IV3年龄小于15岁未行MRI检查)、B超(肝、胆、胰、脾、肾、肾上腺及盆腔)、眼底、血尿常规及尿儿茶酚胺检测,其中3例(1III3、3III1、3III4)携带者检查发现异常。1III3,21岁,腹部CT提示多发胰腺囊肿,见图3D。3III1,33岁,平日偶感腹胀,未予重视,此次基因检查突变阳性后行腹部CT发现胰腺及双肾多发囊肿,见图3E。3III4,22岁,基因检测阳性情况下眼底检查发现双侧眼底视网膜血管母细胞瘤,造影见瘤体及粗大扭曲的滋养血管,见图3F。
3个家族共14例发病患者,男女发病比例4∶3,其中3例患者经基因筛查后临床检查证实已发病。14例发病患者首次出现症状的年龄为19~58(30.2±10.7)岁。病变部位包括中枢神经系统血管网状细胞瘤6例、肾透明细胞癌5例、视网膜血管母细胞瘤2例、胰腺多发囊肿6例、肾脏多发囊肿8例,嗜铬细胞瘤5例。中枢神经系统血管母细胞瘤患者除家系3中3II5未手术即死亡外,余患者共7次小脑肿瘤手术,肿瘤平均复发时间5.8年。5例肾透明细胞癌的患者,仅1例患者7年内连续行4次肾脏手术,肿瘤平均复发时间1.75年,余4例均因发现晚而延误治疗。3个家族共5例患者死亡,死亡率35.7%。2例死于中枢神经系统血管母细胞瘤,3例死于肾癌,平均死亡年龄33岁。
3 讨论
VHL综合征是常染色体显性遗传病,因vhl肿瘤抑癌基因突变所致,遗传率50%,该抑癌基因包含3个外显子,共转录213个氨基酸。外显子2的表达(亚型I)或不表达(亚型II)导致基因转录为2种不同的剪接RNA。亚型I有2种转录结果:pVHL30和pVHL19,两者在人体组织中均广泛表达并扮演着控制转录伸展率的角色,二者统称为pVHL。低氧诱导因子HIF1和HIF2是DNA结合转录因子,掌控基因转录激活促进细胞在低氧环境下适应及生长。pVHL可与elongin B、elongin C、cullin-2及Rbx1等其他蛋白结合形成复合物VCB-Cu12,
继而作用于HIF1和HIF2ɑ亚基来调节泛素介导的蛋白降解。低氧的环境或pVHL丧失及活性下降时,HIF1和HIF2稳定并成为低氧反应的转录因子。这种低氧反应影响了葡萄糖的摄取和代谢,使血管生成,形成细胞外基质和细胞增殖,从而涉及VHL综合征相关肿瘤的产生[3-6]。

表3 3个家族15例基因检测阳性者临床发病特点
张进等[7]完成了中国9个VHL家族的基因突变研究,其中错义突变最多(7/9),突变的位置主要集中在第1外显子的后1/3。有学者对中国各地共70个家族基因检测结果进行总结,发现点突变80%,大片段缺失17.3%,18个突变位于外显子1 (26个家族),7个突变位于外显子2(7个家族),11个突变位于外显子3(18个家族),1个突变位于外显子2,1个突变位于3’-UTR。可见外显子1和外显子3为突变高发区域(51%)[8]。本组研究显示第1、3家族均为点突变,家族突变位点均位于外显子1后2/3,另1个家族外显子3碱基C缺失。
临床上VHL综合征多采用Maher et al[2]提出的诊断标准:①有家族史,患中枢神经系统血管母细胞瘤或视网膜血管母细胞瘤、肾细胞癌、嗜铬细胞瘤以及内囊淋巴瘤其中任一疾病,建立诊断。②无家族史,≥2种中枢神经系统血管母细胞瘤,或者有1种中枢神经系统血管母细胞瘤合并一处内脏病变建立诊断。本组3个先证者有明确家族史,已临床诊断VHL综合征。家族成员1Ⅲ3、3Ⅲ1、3Ⅲ4、3Ⅳ3,基因筛查阳性,临床检查表明3例家族成员已发病。前2例患者表现为胰腺和肾脏囊肿,可能为胰腺神经内分泌肿瘤及肾透明细胞癌的癌前病变,予之随访观察。患者3Ⅲ4表现为视网膜血管母细胞瘤,为防止病灶逐渐扩大及相关并发症影响视力,2014年7月在临床指导下行激光治疗。
研究[2]显示,vhl基因型与临床表现型存在相关性,基因错义突变所编码的蛋白常表现为嗜铬细胞瘤。本组家族1因外显子1第330位碱基A替换为
碱基C,发生错义突变,家族3因外显子1碱基A转换为碱基G,导致错义突变。但是同一家族之间可见发病的时间与病变累及部位不尽相同。如家族2,先证者与其孪生哥哥,都有肾脏病变及嗜铬细胞瘤,但先证者未出现中枢神经系统血管母细胞瘤。
临床调查表明VHL综合征具有发病年龄轻、多呈隐匿发病、肿瘤易复发、死亡率高、患者生存时间短的特点。本组14例发病患者,首次出现症状的平均年龄为30.2岁,5例患者出现症状后无法手术治疗。5例中枢神经系统血管母细胞瘤患者共7次小脑肿瘤手术,肿瘤平均复发时间5.8年。1例肾透明细胞癌患者7年内连续行4次肾脏手术,肿瘤平均复发时间1.75年。5例患者死亡,死亡率35.7%,平均死亡年龄33岁。
VHL综合征表现为全身多系统病变,病变部分早期隐匿,临床上难以早期诊断,导致患者治疗效果不佳,死亡率高。该疾病在患者家族引起恐慌,基因检测可明确诊断并筛选出基因突变携带者,消除家族成员恐慌情绪。对基因突变阳性成员进行临床随诊,可以把握患者治疗时机、提高治疗效果、延长生存时间及提高生活质量。
参考文献
[1] Maher E R,Iselius L,Yates J R,et al.von Hippel-Lindau disease:a geneticstudy[J].Med Genet,1991,28(7):443-7.
[2] Maher E R,Yates J R,Harries R,et al.Clinical features and natural history of von Hippel-Lindau disease[J].Q J Med,1990,77(283):1151-63.
[3] Richards F M,Phipps M E,Latif F,et al.Mapping the von Hippel-Lindaudisease tumour suppressor gene:identification ofgermline deletions by pulsed field gel electrophoresis[J].Hum Mol Genet,1993,2(7):879-82.
[4] McNeill A,Rattenberry E,Barber R,et al.Genotype-phenotype correlationsin VHL exon deletions[J].Am J Med Genet A,2009,149A(10):2147-51.
[5] Richards F M,Schofield P N,Fleming S,et al.Expression of the von Hippel-Lindau disease tumour suppressor gene during humanembryogenesis[J].Hum Mol Genet,1996,5(5):639-44.
[6] Kaelin W G Jr.The von Hippel-Lindau tumour suppressor protein:O2sensing and cancer[J],Nat Rev Cancer,2008,8(1):865-73.
[7] 张 进,黄翼然,潘家骅,等.中国人von Hippel-Lindau综合征种系突变研究[J].中华医学遗传学杂志,2007,24(2):124-7.
[8] Wu P,Zhang N,Wang X,et al.Family history of von Hippel-Lindau disease was uncommon in Chinese patients:suggesting the higher frequency of de novo mutations in VHL gene in these patients[J].J Hum Genet,2012,57(4):238-43.
Mutation analysis and clinical investigation of three Chinese families with VHL disease
Wu Xinyao1,Chen Jiangming2,Zhao Yijun1,et al
(1Organ Transplantation Center,The First Affiliated Hospital of Anhui Medical University,Hefei 230022;2Dept of General Surgery,The Second Affiliated Hospital of Anhui Medical University,Hefei 230601)
AbstractObjective To detect the genetic mutations of vhl gene of three families who were diagnosed in clinical,and screen the carriers for clinical guiding treatment.Methods The vhl gene was screened for mutation using a direct DNA sequencing analysis for 31 members from three families.The genetic mutations carriers were required to undergo further clinical examination,surveillance and treatment.Results The genetic testing results were based on positive three probands.5 members of family 1 had positive genes,1 member in family 2,7 members in family 3.The position of three von Hippel-Lindau syndrome(VHL)families genes mutations sites was separately on c.330C>A in exon1,488delC in exon3 and c.233G>A in exon1.In DNA sequencing analysis,we found four potential carriers(1Ⅲ3,3Ⅲ1,3Ⅲ4,3Ⅳ3),and combined with clinical examination,three(1Ⅲ3,3Ⅲ1,3Ⅳ3)of them had disease.1Ⅲ3 was diagnosed with multi-cysts on kidney and pancreas,3Ⅲ1 was diagnosed with pancreatic multi-cysts.3Ⅲ4 was diagnosed with retinal hemangioblastoma.We had guided the member 3IV3 for eye surgery.3IV3 was not found with abnormal symptoms because of her young age.And all of the gene carriers should have the follow-up visit.Conclusion Genetic testing can diagnose VHL syndrome early.To improve patients'treatment,prolong survival and improve the quality of life,the patients with vhl gene mutations need follow-up closely.
Key wordsvon Hippel-Lindau syndrome;vhl gene;gene detection
作者简介:吴鑫尧,男,硕士研究生;刘付宝,男,副教授,副主任医师,硕士生导师,责任作者,E-mail:liufubao88@163.com
基金项目:安徽省自然科学基金(编号:1308085MH133)
文献标志码A
文章编号1000-1492(2015)05-0635-05
中图分类号R 394.3
