2013年度江苏省药品生产企业新版GMP认证检查缺陷项目分析
2014-04-30王金伟陈永飞
王金伟,陈永飞
江苏省食品药品监督管理局认证审评中心,南京 210002
2013年度江苏省药品生产企业新版GMP认证检查缺陷项目分析
王金伟,陈永飞
江苏省食品药品监督管理局认证审评中心,南京 210002
对江苏省药品生产企业在2013年新版药品GMP认证检查中存在的缺陷项目进行分析,为药品监管部门的认证检查、日常监管和药品生产企业完善GMP工作提供参考。
GMP;认证检查;缺陷项目
《药品生产质量管理规范(2010年修订)》(简称新版GMP)是适用于制药行业的强制性标准。据国家食品药品监督管理局对药品生产企业的要求,现有药品生产企业血液制品、疫苗、注射剂等无菌药品的生产,应在2013年12月31日前达到新版GMP要求,其他类别药品的生产企业均应在2015年12月31日前达到要求。我省自2011年起实施新版GMP认证。现对2013年新版GMP认证检查中发现的缺陷构成情况及高频次出现的缺陷进行分析,找出企业在实施GMP过程中存在的共性问题,并提出相应的对策。
1 总体情况
2013年,我中心共对126家企业进行了155次新版GMP认证,现场检查通过率为100%,检查组共发现缺陷1462条,其中严重缺陷0项,主要缺陷31条,一般缺陷1431条,详见图1。《药品生产质量管理规范认证管理办法》规定:严重缺陷指与药品GMP要求有严重偏离,产品可能对使用者造成危害的缺陷;主要缺陷指与药品GMP要求有较大偏离的缺陷;一般缺陷指偏离药品GMP要求,但尚未达到严重缺陷和主要缺陷程度的缺陷。
2 主要缺陷情况分析
2.1 概述

图1 认证检查缺陷项目分布图
31条主要缺陷涉及24家企业,占申请GMP认证企业数的19%。6条主要缺陷出现在5家中药饮片生产企业,占17家饮片认证企业的29.4%。2条主要缺陷出现在2家医用氧生产企业,占5家医用氧认证企业的40%。9条主要缺陷出现在8家原料药生产企业,占73家原料药认证企业的11.0%。中药饮片产企业、医用氧、原料药出现主要缺陷的比例显著高于平均水平。
31条主要缺陷中出现频次最高的是确认与验证,共7条,占主要缺陷的22.6%;其次,机构与人员、文件管理,各6条,各占19.4%;还有厂房与设施、质量控制与质量保证均为4条,占12.9%。
2.2 确认与验证方面缺陷
企业在确认与验证方面的主要缺陷表现在两个方面:①关键设备确认、清洁验证或是工艺验证工作没有做;②确认或验证的项目和内容不全,数据不完整。该类别缺陷占主要缺陷总数的比例为22.6%,相较2012年13.8%的数据,呈递增的趋势,需要引起药品监管部门的注意。究其原因,仍是企业重视不够,特别是人员资质不高、管理水平较低的企业,没有认识到确认与验证对于确保持续稳定生产出符合要求产品的重要性。见表1。需要说明的是表1中“缺陷项目”指的是不符合新版GMP具体条款要求的缺陷编号。

表1 确认与验证主要缺陷项目
2.3 机构与人员方面缺陷
企业在机构与人员方面的主要缺陷表现在三个方面:①培训效果较差;②人员资质不符合要求;③人员配置不足,个人承担的职责过多。

表2 机构与人员主要缺陷项目
从表2还可以看出,6条主要缺陷中,中药饮片和原料药、制剂生产企业各出现2条,但结合2013年度相关生产企业认证数,可以得出两个结论:①中药饮片生产企业依然轻视培训工作,普遍存在培训针对性不强和培训效果较差的情况;②个别原料药、制剂企业由于经济效益差,关键人员资质不符合要求,人员配置不齐,且不将培训工作落到实处,在1998版药品GMP证书到期前抱着侥幸心理申请2010版GMP认证,怀有“闯关”的心态,这一动向需要引起药品监管部门的警惕。
2.4 文件管理方面缺陷
企业在文件管理方面的主要缺陷表现在两个方面:①批生产记录设计的内容不完整,未能根据工艺规程合理设计,缺少相关操作内容;②工艺规程内容不全,缺少工艺参数,或者工艺规程与现行其他文件要求不一致。从表3可以看出,中药饮片生产企业在文件管理方面普遍存在此类问题。此外,中药提取物、中药制剂的生产企业在文件管理方面易出现主要缺陷。

表3 文件管理主要缺陷项目
2.5 厂房与设施方面缺陷
企业在厂房与设施方面的主要缺陷表现在三个方面:①厂房、设施、设备没有合理设计与布局,不能有效防止污染、交叉污染、混淆和差错;②设施选择不合理,不便于清洁;③产尘操作间缺少除尘设施。
从表4可以看出:①个别原料药、无菌原料药生产企业忽视设施应便于清洁的要求,不重视产尘操作间的除尘设计和除尘效果;②部分医用氧生产企业原先是生产工业氧的,缺乏GMP理念,拼凑上马,厂房未能合理布局和改建;③个别生产含激素类制剂的企业,在清洁验证不充分的前提下,缺少专门取样、称量功能间,不能有效避免交叉污染。

表4 厂房与设施主要缺陷项目
2.6 质量控制与质量保证方面缺陷
企业在质量控制与质量保证方面的主要缺陷表现在三个方面:①物料和产品检验不符合要求,表现为部分检验方法未确认和检验原始记录不全;②取样管理不规范,如使用未经处理的样品容器而影响检验结果;③标准品、对照品管理不规范,多次使用的自制对照品未通过定期标化证明其效价或含量在有效期内的稳定,从而影响检验结果,风险很高。见表5。
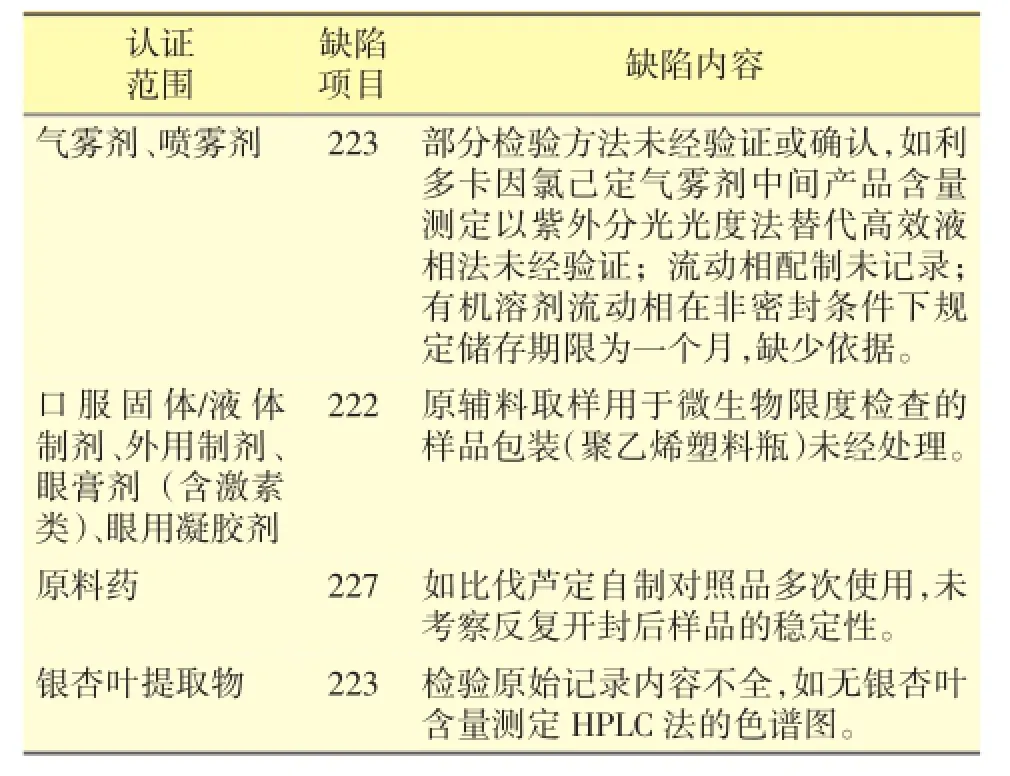
表5 质量控制与质量保证主要缺陷项目
需要说明的是,对于新取得药品批准文号就申请GMP认证的原料药,现场检查时会关注其自制工作标准品或对照品的管理,企业应完善相关质量标准以及制备、鉴别、检验、批准和贮存的操作规程,建立健全标化记录。
3 一般缺陷情况分析
3.1 概述
1431条一般缺陷中设备管理方面共265条,占缺陷总数的18.5%,见表6;文件管理方面共252条,占缺陷总数的17.6%,见表7;质量控制与质量保证方面共231条,占缺陷总数的16.1%,见表8。
和2012年的一般缺陷相比,上述设备、文件管理、质量控制与质量保证缺陷类别的位次排名有些变化,但所占缺陷总数的比例变化不大,根本原因依然是:①企业没有认识到设备在GMP实施中扮演的重要角色,同时国内制药设备的制造水平不能满足制药企业的发展需求,如设备的设计、材质、精度、易于清洗等方面;②企业忽视文件管理对于质量保证体系的重要性,文件缺乏详细内容和可操作性,不符合生产实际需求;③企业未认真学习新版GMP关于质量控制实验室管理方面的规定,相关工作不符合规范要求。

表6 设备管理一般缺陷项目

表7 文件管理一般缺陷项目

表8 质量控制与质量保证一般缺陷项目
3.2 设备管理方面缺陷
企业在设备管理方面的一般缺陷表现在五个方面:①设备预防维护工作不到位,故障设备没有维修,如某厂片剂车间包衣降尘处理装置、室外管道未及时维护;原料药综合车间洁净区离心机内部锈蚀,未针对性制定预防维护计划。②设备和检验仪器使用日志管理不符合要求,如药品生产设备使用日志未记录药品规格、批号,部分设备如料斗清洗机、称量工序未建立使用日志。③设备清洁不彻底或者清洁规程内容不符合要求,如某厂固体制剂车间已清洁的万能高速粉碎机有物料残留,清洁规程未规定设备可拆卸部件的清洗要求。④现有设备存在不方便操作、清洁、维护的问题,如某厂原料车间进入洁净区的物料管道不便于清洗,制粒干燥工序输送泵使用的密封材料为生胶带,选材不合理。⑤校准管理不符合要求,存在缺少校验标识、校验超过有效期、校准的量程范围未涵盖实际生产的使用范围。以上看出,人为因素造成的缺陷占大多数,说明员工的责任心和规范操作的意识不强,仍须加强培训和管理。
3.3 文件管理方面缺陷
企业在文件管理方面的一般缺陷表现为:未根据工艺规程或结合实际生产操作来设计批生产记录,记录缺少部分内容,文字不确切、不清晰、不易懂,可操作性较差,如空调系统操作规程未规定初、中效过滤器更换后记录初始压差值;压片工序片重差异QA的确认过程、粘合剂制备用搅拌勺、打浆桶等器具的干燥过程缺少记录。这是很多企业普遍存在的问题,应高度重视文件对生产的指导性地位,清醒地认识到文件是整个质量管理体系的基础,也是自检、审计和认证检查的依据,结合生产实际对相关文件进行梳理,避免出现文件和操作两张皮的现象,真正做到“记我所做的”、“做我所写的”。
3.4 质量控制与质量保证方面缺陷
企业在质量控制与质量保证方面的一般缺陷集中在药品检验,表现为四个方面:①试剂、试液、培养基和检定菌的管理不规范,如缺少大肠埃希菌菌种甘油管的制备及数量、菌种复苏、操作人、使用数量等信息,样品分样和稳定性考察未建立台账。②检验记录内容不全或记录不规范,如纯化水电导率测定记录缺少仪器型号、编号、温度,不挥发物缺少恒重过程,缺少检验环境的温湿度记录。③取样管理不规范,如取样人员未经授权,取样标准操作规程未规定取样器具、存放样品容器的类型和状态、取样后剩余部分及样品的处置和标识等。④标准品、对照品管理不规范,如未按规定贮存,缺少适当的标识。
4 结论及建议
2013年新版药品GMP认证的平均缺陷数为9.43条,平均每次检查发现的缺陷数少于10条,和新版药品GMP实施前10条左右的数据相差不大,且远低于美国FDA或者欧盟药监部门对我省企业认证检查时发现的平均缺陷数。
新版药品GMP和1998版相比,内容上有很大丰富,仅从通则的具体条款上来看,新版药品GMP为312条,1998版GMP为85条,条款数增加将近3倍。按常理推断,当规范的标准提高和内容更具体时,发现的问题应该更多,但结果是基本持平,甚至略少。
针对此问题,排除企业认真实施GMP的原因外,还有三个方面值得思考:①检查员自身须加强学习,提高业务水平,增强发现问题和分析问题能力。②现场检查中发现的问题应全部体现在检查报告中,即使企业针对现场检查发现的问题及时整改的,也应该在检查报告中详细说明。③药品监管部门、检查员和企业都应转变理念,不要以缺陷数的多少来评估GMP实施水平,要依据药品质量风险管理从整体质量体系上来判断是否符合新版药品GMP的要求。
Analysis on the Instance of Revised Drug GMP Certification Inspection for Jiangsu Province in 2013
WAN Jing-wei,CHEN Yong-fei
Jiangsu Center for Drug Evaluation & Certification,Nanjing 210002
In this article,we analyzed the defective items observed in revised drug GMP certification inspection of pharmaceutical manufactures for Jiangsu Province in 2013.This research aims to provide reference for certification inspection,daily supervision and improving GMP work.
GMP;certification inspection;defective items
R951
A
1673-7806(2014)06-574-04
王金伟,男,主管中药师 E-mail:13951882291@139.com
2014-03-12
2014-07-31
