布洛芬、右旋布洛芬及其衍生物的合成进展
2010-12-08汪家华郑土才王
汪家华 郑土才王 平
(1.浙江巨化集团公司制药厂,浙江 衢州 324000;2.浙江工业大学浙西分校化学与制药工程系,浙江 衢州324000;3.浙江工业大学药学院,杭州310014)
综 述
布洛芬、右旋布洛芬及其衍生物的合成进展
汪家华1郑土才2王 平3
(1.浙江巨化集团公司制药厂,浙江 衢州 324000;2.浙江工业大学浙西分校化学与制药工程系,浙江 衢州324000;3.浙江工业大学药学院,杭州310014)
本文先评述了转位重排、醇羰基化、烯烃羰基化、卤代烃羰基化、烯烃催化加氢和环氧丙烷重排等6种布洛芬的合成方法,然后总结了布洛芬衍生物如精氨酸盐、各种酯、酰胺等的合成及主要特点,并简要回顾了右旋布洛芬及其衍生物的合成进展等,最后认为转位重排仍然是最适合我国实际的生产方法,分析了该方法的关键技术要点,并提出应加快右旋布洛芬和副作用更小的布洛芬衍生物的开发。
布洛芬;右旋布洛芬;羰基化;转位重排;拆分;合成
布洛芬(ibuprofen),化学名为2-(4-异丁基苯基)丙酸,为新一代非甾体消炎镇痛药物,具有比阿司匹林更强的解热、消炎和镇痛作用,副作用则比阿司匹林小得多。因此自上世纪70年代末上市以来,获得了迅速发展,现已成为生产量和使用量最大的消炎解热镇痛药之一,目前全球的年产量已超万吨。有关其工艺改进和新工艺、拆分或不对称合成获得其手性体、其衍生物以及各种制剂的研究报道层出不穷[1-2]。笔者对近年来布洛芬及其衍生物和右旋布洛芬及其衍生物的合成进展作一综述。
1 布洛芬的合成
1.1 转位重排法
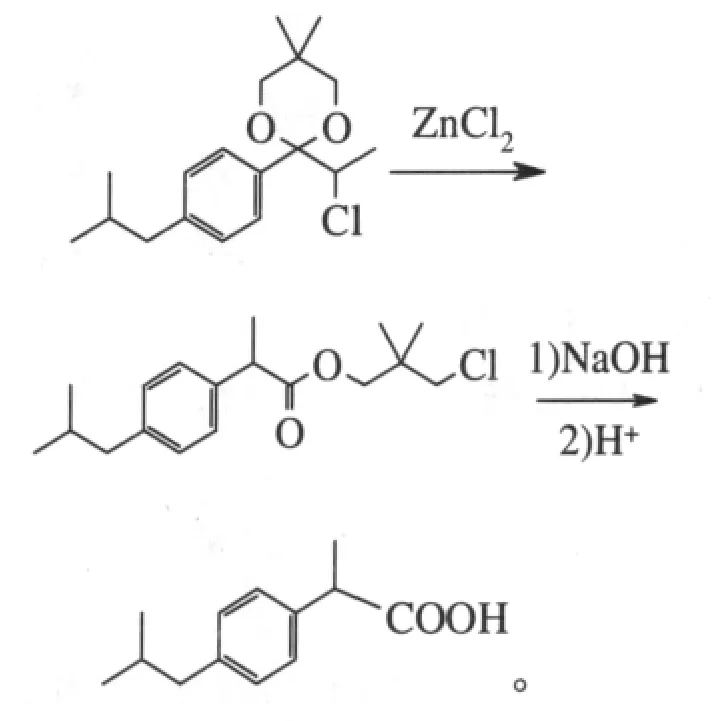
芳基1,2-转位重排法是目前国内厂家普遍采用的一种合成方法,它以异丁苯为原料,经与2-氯丙酰氯的傅克酰化、与新戊二醇的催化缩酮化、催化重排、水解等制得布洛芬[1]。反应式为:
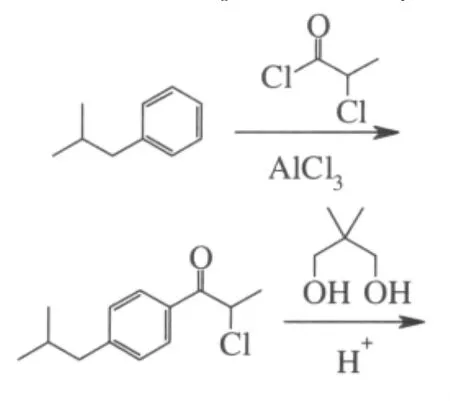
李兴泰等对傅克酰化进行了研究,以无水三氯化铝与2-氯丙酰氯在无溶剂条件下20~40℃反应产生活性络合物,然后15~30℃滴加异丁苯进行酰化,反应结束再加入石油醚,便于分层、水洗及下一步缩酮时带水[3]。该工艺的优点为:避免了酰化时以石油醚作溶剂其中所含微量芳烃杂质所带来的副产物,或使用二氯乙烷作溶剂时所带来的毒性和溶剂残留问题,避免了传统工艺使用冷冻盐水的要求,降低了能耗和设备腐蚀等。
石文平等将固体超强酸SO42-/Fe2O3-ZrO2-La2O3用于缩酮化反应,代替传统的硫酸或对甲苯磺酸,取得了较好的效果[4]。作者给出了催化剂的最佳制备条件和缩酮反应的最佳条件。催化剂不仅活性很高,而且重复使用性能良好。
舒瑞友对重排反应进行了研究,以布洛芬锌替代原工艺中的氧化锌为催化剂,使重排反应成为均相液体回流反应,反应温度降低,时间缩短,操作更简单稳定,粗品布洛芬颜色为类白色,同时还报道了布洛芬锌的制备方法[5]。
1.2 醇羰基化法
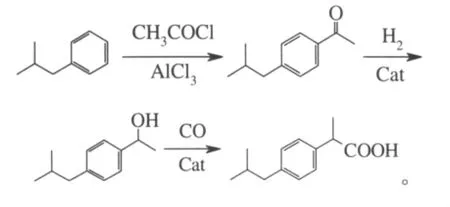
醇羰基化法即BHC法,以异丁苯为原料,经与乙酰氯的傅克酰化、催化加氢还原和催化羰基化3步反应制得布洛芬,为目前最先进的工艺路线,为国外多数厂家所采用。
Manimaran等对经典酰化工艺进行了详尽的研究,发现在很低温度下如0、-10℃,甚至-35℃酰化仍很容易进行,但产生的异构体大为减少[6]。例如,乙酰氯和异丁苯冷至-30℃以下,少量多批加入无水三氯化铝,保持-30℃以下加料和反应,冰解后产物的GC分析显示各物质的质量分数分别为:对异丁基苯乙酮98.8%,间位异构体0.8%,其他高沸杂质0.4%。
Chaudhari等报道了羰基化反应在卤离子源、质子酸、水和具有合适催化剂及有机溶剂存在下,与CO在2相或均相状态下反应[7]:
其中羰基化催化剂的中心金属为钯或铂,主要配体为8-羟基喹啉、2-羟基吡啶、2-(2-羟乙基)吡啶、吡啶-2-甲酸、哌啶-2-甲酸、喹啉-2-甲酸、异喹啉-1-甲酸和异喹啉-3-甲酸。此前文献报道所用催化剂为 Pd(PPh3)2Cl2、PdCl2或 Pd(OAc)2和过量膦配体,反应速率低,选择性差。Chaudhari等报道的催化体系具有反应速率快,即使在较低压力下也有很高的选择性,催化剂回收简单高效等优点,避免使用过量配体,反应可在2相也可在均相条件下温和地进行。
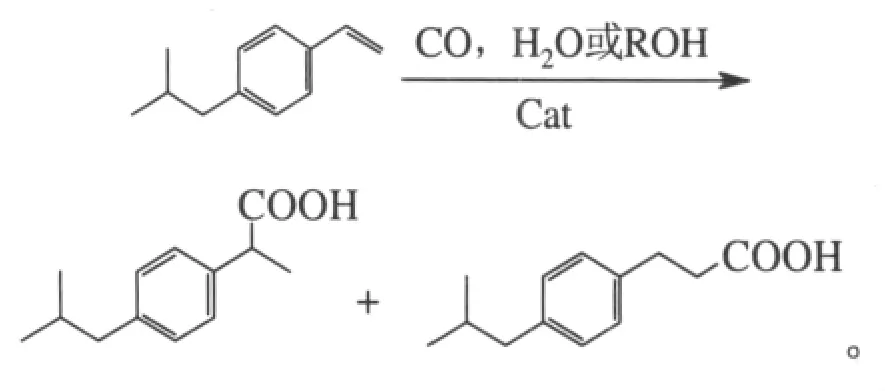
1.3 烯烃羰基化法
早有报道芳基取代烯烃与CO和水或醇在钯催化剂和酸性条件下生成芳烷基羧酸或羧酸酯。Wu报道了无氧条件下钯的催化活性可通过与某些配体的合用而增强[8]。例如,在相同条件下,1:1的n(Ph3P):n(Ph3PO)与氯化钯合用时效果最佳,不仅反应速率快,转化率最高,而且几乎没有异构体3-(4-异丁基苯基)丙酸的产生。反应式如下:
1.4 卤代烃羰基化法
卤代烃羰基化法以1-对异丁基苯基-1-氯乙烷为原料,经与CO在催化剂和碱性条件下羰基化生成产物,反应式如下:

该方法在上世纪80年代即有报道,催化剂一般为钴或钯的化合物,溶剂为醇类。但有如下缺点:碱性条件下得到的一般为布洛芬盐,需要增加酸化一步才能得到布洛芬;卤代烃羰基化往往产生双羰基化副产物,即4-异丁基苯基丙酮酸;具有良好选择性的反应参数的变化范围很窄。Elango报道了以钯为催化剂的酸性水溶液中的羰基化反应,同时还报道了异丁苯与乙醛和氯化氢进行氯乙基化反应生成1-对异丁基苯基-1-氯乙烷的详细操作[9]。
典型的氯乙基化工艺如下:异丁苯(3 mol)和氯化锌(1 mol)加入1 L反应瓶中,10℃以下2 h内滴入乙醛(1 mol)与异丁苯(0.5 mol)的混合物;将反应液加热至室温,向反应物中通氯化氢气体约2 h,继续反应6 h;反应液加水、分层,碳酸氢钠洗、水洗、干燥,蒸馏回收过量异丁苯,真空蒸馏得产物。
羰基化使用的催化剂如PdCl2(PPh3)2,羰基化反应转化率较高,但布洛芬的选择性最高仅有74%,主要副产物为异构体和聚合的重组分。
1.5 烯烃催化加氢法
Chan和Pai报道了利用手性配体的钌配合物催化2-(6-甲氧基-2-萘基)丙烯酸的加氢制备萘普生,对映体过量(ee)达96%[10]。反应式如下:
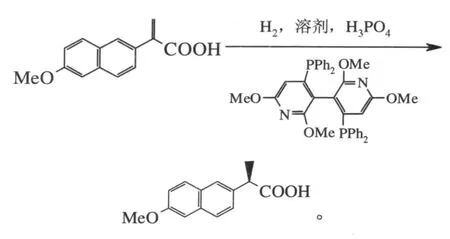
也可用于2-(4-异丁基苯基)丙烯酸的催化加氢。作者主要研究了手性配体和催化剂的制备、催化剂的回收、氢气压力、溶剂、添加磷酸和反应温度等对反应产物光学纯度的影响。具体的手性配体为Figure 5中的3,3'-联吡啶双膦化合物。
姜标等报道了上述反应的前体化合物2-芳基乳酸酯的制备方法[11]。反应式如下:

重点研究了芳香烃包括异丁苯与丙酮酸甲酯或乙酯在Lewis酸催化下缩合生成2-芳基乳酸酯的条件,包括催化剂种类、用量、反应溶剂、反应温度等,但反应要求低温如-30℃,并且收率中等。
1.6 环氧丙烷重排法
陈平等报道了一种新的布洛芬合成方法,其中制备对异丁基苯乙酮及由2-(4-异丁基苯基)丙醛转化为布洛芬的2步反应与经典的Darzens缩合法的相应步骤相同[12]。反应式如下:
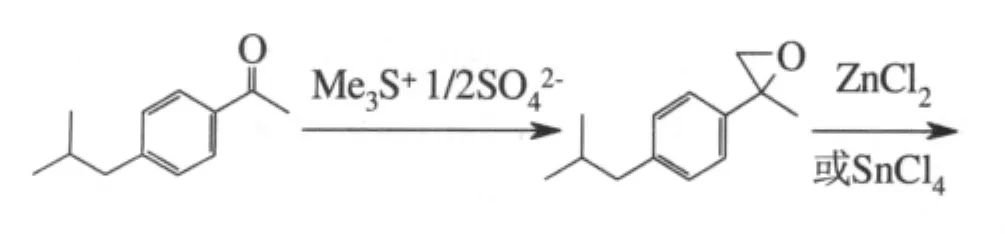
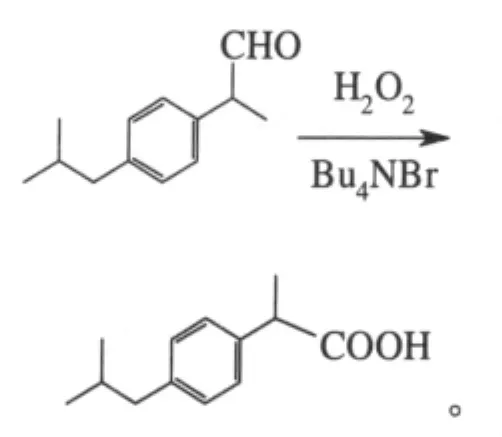
对异丁基苯乙酮与硫叶立德(由二甲硫醚与硫酸二甲酯反应制得)反应得2-(对异丁基苯基)-1,2-环氧丙烷,然后以无水氯化锌或无水氯化锡催化重排得2-(对异丁基苯基)丙醛。专利还改进了中间体醛氧化制布洛芬的条件,以四丁基溴化铵为相转移催化剂,30%过氧化氢为氧化剂,收率近90%。
综合分析以上不同的工艺路线,笔者认为仍以经典的1,2-转位重排法比较适合国内生产。醇羰基化法和烯烃羰基化法技术难度高,催化剂价昂;卤代烃羰基化法选择性低,催化剂价高,氯乙基化时易发生多烷基化和双芳基化,杂质多;烯烃加氢法虽有可分别制备消旋布洛芬和右旋布洛芬的优点,但2-(4-异丁基苯基)丙烯酸需由异丁苯经2步反应制得,与丙酮酸酯缩合一步收率低,丙酮酸酯价格高,加氢催化剂昂贵;环氧丙烷重排法有4步反应,二甲硫醚污染大,工业化前景不乐观。
1,2-转位重排法虽也为4步反应,但每步收率高、安全性好、重排催化剂价廉易得且无毒。2-氯丙酰氯的供应充足,价格低廉,为该合成方法的稳定运行提供了充分的保障。本工艺的关键是酰化时减少副产物的产生,提高溶剂回收率,缩酮时最大限度提高转化率,重排平稳,回收或利用重排时副产的3-氯-2,2-二甲基-1-丙醇,此外若能将酰化的催化剂三氯化铝改为固体酸,则不仅能降低成本,还能减少设备腐蚀,提高操作安全性。
2 布洛芬衍生物的合成
Akbarali等报道了制备布洛芬钠盐二水合物的1种方法:将2-乙基己酸钠的双蒸水溶液滴加入布洛芬的四氢呋喃(THF)溶液中,控温28℃,搅拌6 h;过滤,完全蒸去THF后得糖浆状液体,将其缓慢倾入大量丙酮中并搅拌1 h,过滤,丙酮洗涤,抽干;45~50℃真空干燥得到产品,HPLC分析目标物质量分数99%,水的质量分数13.0%[13]。
董雪玲等报道了布洛芬L-精氨酸盐的制备方法:控制一定温度下,将布洛芬溶于体积分数95%乙醇中,分次加入等摩尔的L-精氨酸,搅拌全溶后,保温0.5 h;降至室温后,反应液缓慢倒入丙酮中,析出白色沉淀,继续搅拌片刻,冰箱中冷却12 h,抽滤;丙酮洗2~3次,抽干;60℃烘至恒量,得白色的布洛芬L-精氨酸盐,收率94.2%,HPLC分析目标物质量分数98.65%[14]。
刘杏敏也报道了制备布洛芬精氨酸盐的1种方法:布洛芬溶于无水乙醇中,室温和搅拌下,将精氨酸逐渐加入该溶液中,之后补加一定量无水乙醇,继续搅拌至溶液透明;继续搅拌一定时间后出现浑浊,再搅拌数小时;过滤,冷无水乙醇洗2次,50℃真空干燥,得白色产品,收率大于90%。该发明的重要特点是:成盐和结晶在一个容器中完成,工艺简捷、收率高、重现性好[15]。
王润玲报道了另一种制备方法:将布洛芬溶于体积分数95%乙醇中,搅拌下将L-精氨酸加入该溶液中,加热至70℃,搅拌反应0.5 h后,室温冷却析晶,抽滤,50℃干燥,得产物,收率90.5%,熔点165~167℃。该法的优点是:反应结束后直接从母液中析晶,不必加入其他溶剂稀释;冷却析晶条件为室温,不必冰箱冷却过夜,母液可回收套用[16]。
周金森等首次报道了布洛芬的乙胺和乙二胺盐的制备和表征,并测定了乙胺盐的晶体结构。乙胺盐的制备方法如下:取一定量乙胺水溶液溶于体积分数95%乙醇中,冰水浴冷却下,加入布洛芬,密封搅拌1.5 h,自然挥发溶剂,分别得到布洛芬乙胺盐的无色晶体和白色粉末。室温下,10 mL水溶液能溶解22 g布洛芬乙胺盐,相比布洛芬的溶解度大为提高。乙二胺盐的水溶性则没有明显提高[17]。
宋妮等报道了布洛芬的酰氯与二乙胺基乙醇发生酯化生成布洛芬乙酯
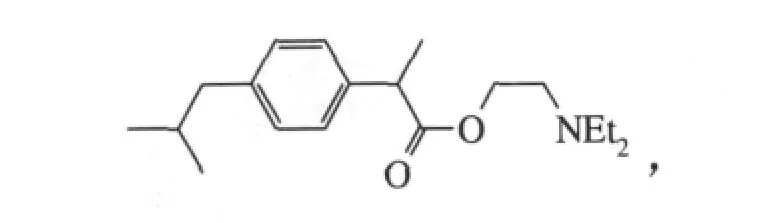
再在丙酮中通入溴甲烷发生季铵化反应合成布洛芬的季铵盐衍生物[18]:
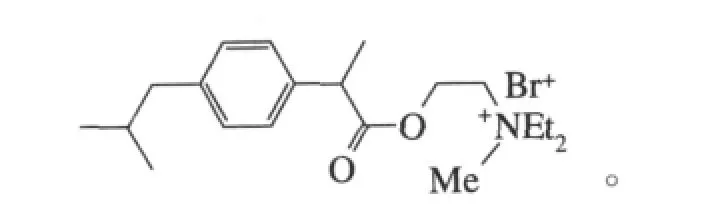
布洛芬乙酯是微生物法拆分消旋布洛芬获取S(+)-布洛芬的前体。刘勇等报道了以HZSM-5分子筛为催化剂,布洛芬与乙醇摩尔比为1.0:1.2,苯为溶剂及带水剂,合成了布洛芬乙酯,收率92%,产物经红外光谱(IR)、气相色谱-质谱(GC-MS)和核磁共振谱(NMR)表征[19]。
向玲等报道了布洛芬经与氯化亚砜反应制成酰氯,再与2-吡啶甲醇缩合制得外用消炎镇痛药布洛芬吡甲酯
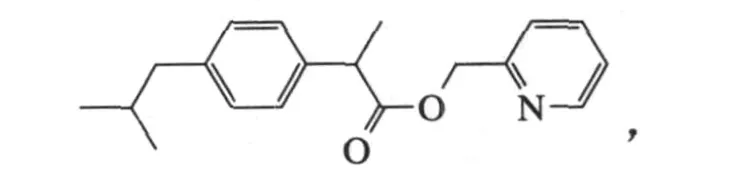
收率93.6%,产物经IR、NMR和MS等表征确证[20]。王鹏报道了布洛芬2-(3,5,6-三甲基)吡嗪酯
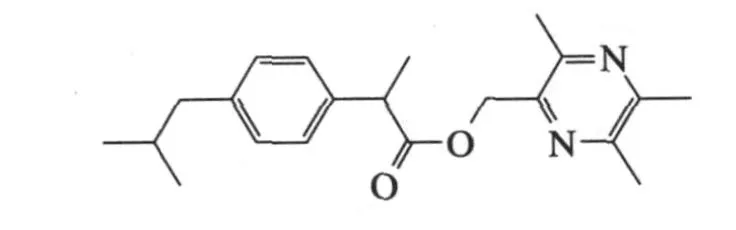
及其盐酸盐的制备,旨在降低布洛芬对胃肠道的刺激性副作用[21]。2,3,5,6-四甲基吡嗪(即川芎嗪)经自由基溴化得2-溴甲基-3,5,6-三甲基吡嗪,再与布洛芬在三乙胺-丙酮中反应得布洛芬2-(3,5,6-三甲基)吡嗪酯,最后成盐。
低相对分子质量药物的高分子化是当前广泛研究的课题。高分子药物一般具有长效、增效、缓释、低毒副作用等优点。孙礼林等报道了布洛芬高分子前体药物及纳米微球的合成和表征,布洛芬经酰氯化后与甲基丙烯酸-2-羟基乙酯酯化生成单体
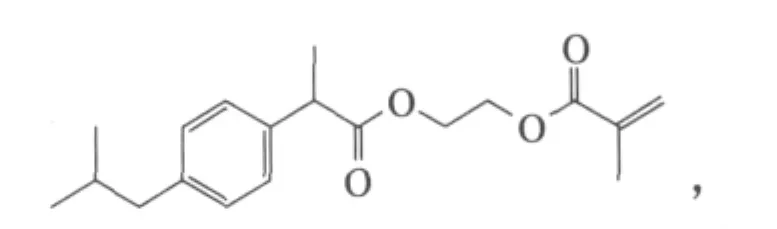
单体分别经聚合或乳液聚合和与甲基丙烯酸甲酯的共聚得到均聚物和共聚物;单体与甲基丙烯酸甲酯共聚还制得了共聚物纳米微球。研究显示,调节单体投料比可以获得不同含药量的高分子药物[22]。
为降低布洛芬羧基引起的胃肠道刺激作用,赵秀丽等研究了布洛芬丁香酚酯
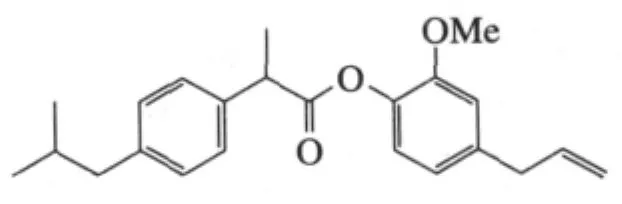
的合成和水解动力学,发现布洛芬丁香酚酯是一个具有良好前景的前体药物,它的合成通过布洛芬的酰氯与丁香酚在无水碳酸钾-丙酮中反应得以实现[23]。
赵一玫等分别将布洛芬与对羟基苯甲酸、水杨酸、对乙酰氨基苯酚(扑热息痛)和5-(2-羟基苯基)-10,15,20-三甲氧基苯基卟啉进行酯化分别得到4种衍生物[24]:
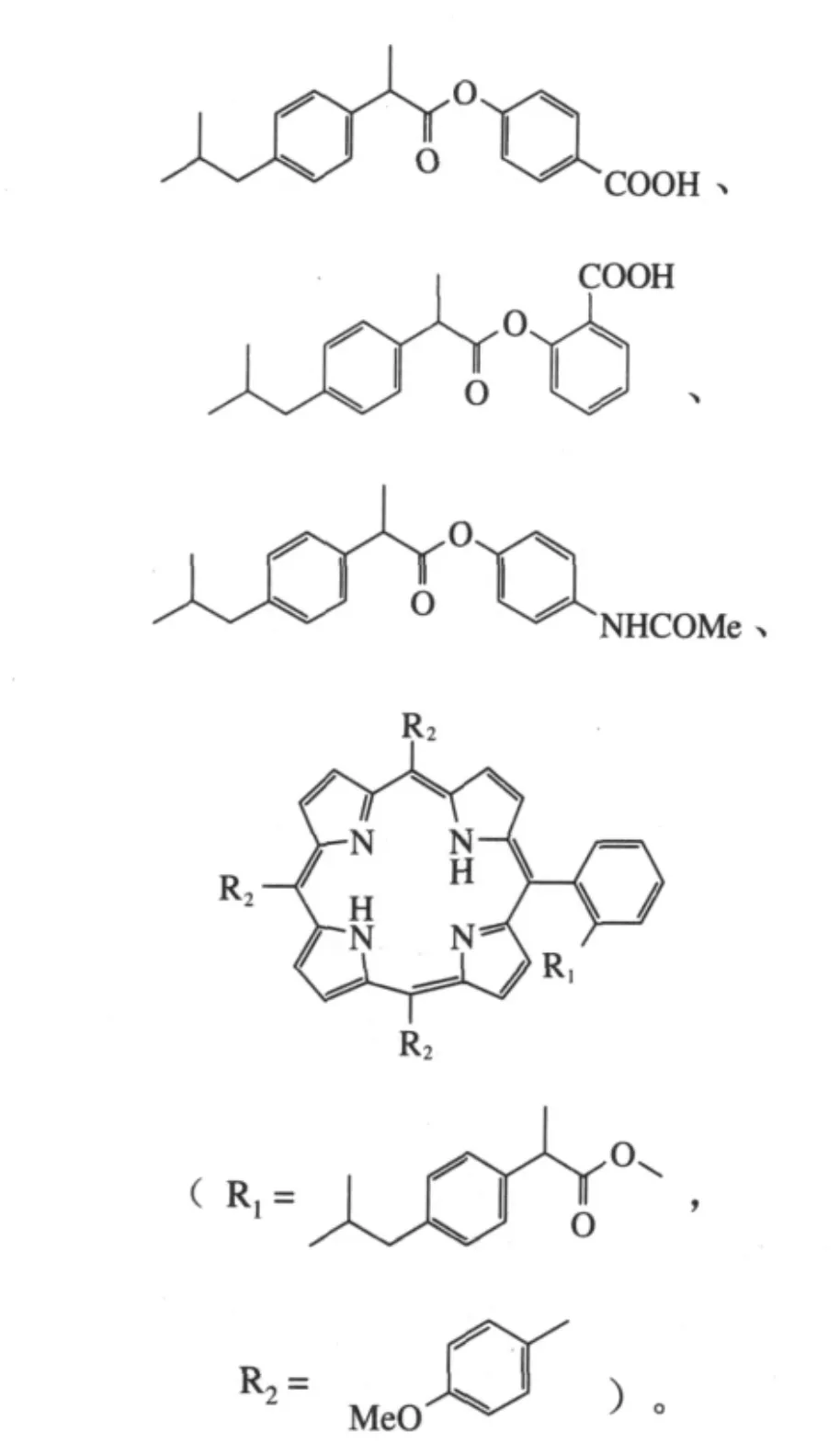
抗炎活性研究表明,第2和第3种的抗炎活性分别是布洛芬的2倍和3倍,值得进一步研究。
魏东芝等利用商品脂肪酶NOVOZYM 435,催化布洛芬和未保护的α-甲基-D-葡萄糖苷的直接酯化制得6-(α-甲基-D-葡萄糖苷)布洛芬
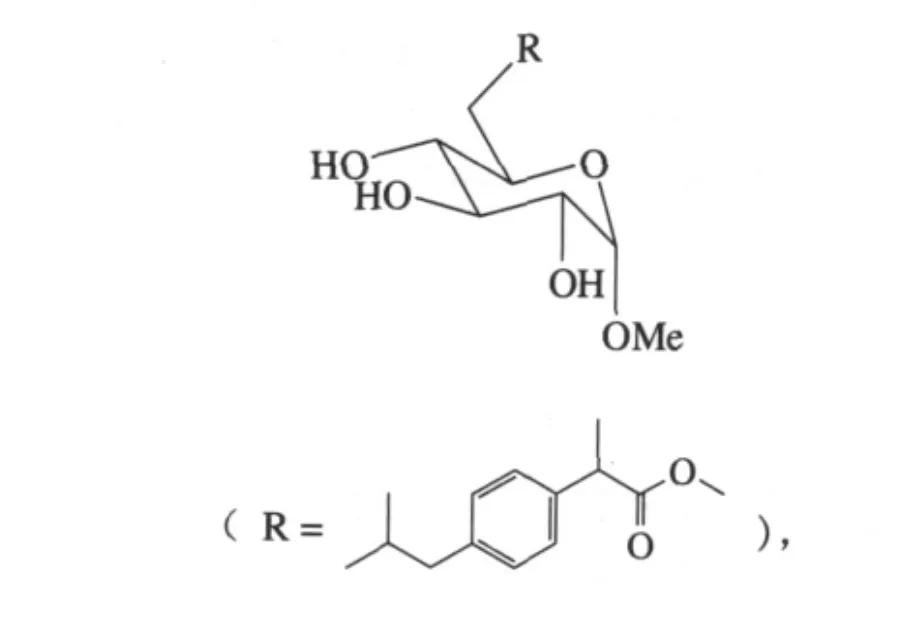
这种布洛芬的糖衍生物的肠胃副作用显著降低,且抗炎活性增强[25]。
王化录研究了广谱抗病毒药三氮唑核苷(利巴韦林)与布洛芬的酰氯在吡啶中反应合成布洛芬三氮唑核苷酯
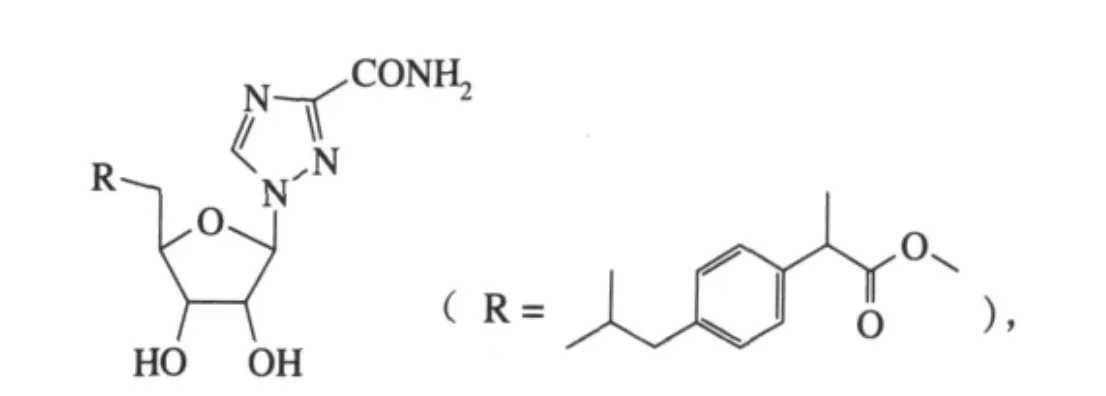
该化合物结合了有解热消炎镇痛作用的布洛芬与有抗病毒作用的三氮唑核苷,研究表明其镇痛、解热、消炎作用与布洛芬相当,而其抗病毒作用则与三氮唑核苷相近,对于由病毒引起的发热疼痛具有很好的应用前景[26]。
宋妮等合成了5个布洛芬糖衍生物,如二缩酮半乳糖的6-位羟基与布洛芬酰氯反应后水解形成的酯
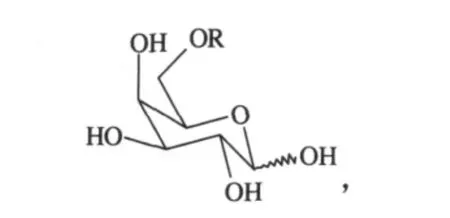
四乙酰基-2-氨基葡萄糖与布洛芬酰氯反应再脱乙酰基形成的酰胺
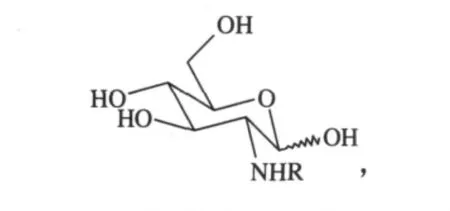
七乙酰基-1-乳糖胺与布洛芬酰氯反应再脱乙酰基形成的酰胺

三乙酰基-2-去氧-2-乙酰胺基葡萄糖胺与布洛芬酰氯反应后经硅胶柱层析得到2个化合物,再分别经脱乙酰化得到1-[2-去氧-2-乙酰胺基-β-D-葡糖氨基]-(-)-布洛芬和1-[2-去氧-2-乙酰胺基-β-D-葡糖氨基]-(+)-布洛芬
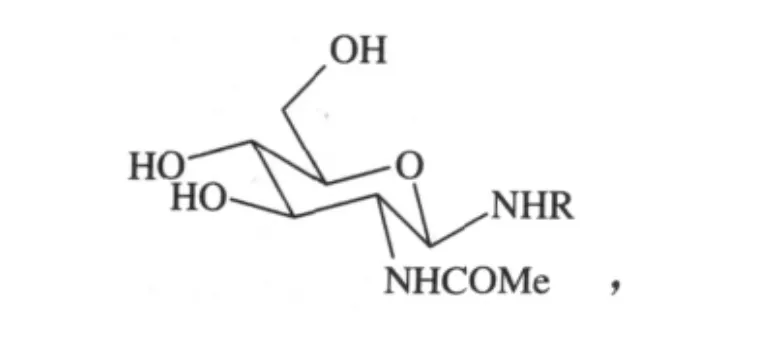
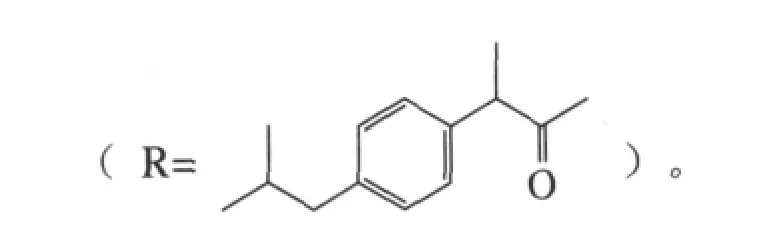
抗炎活性研究表明,第1、第5个(左旋和右旋)的抗炎活性均明显优于布洛芬,其中右旋布洛芬的衍生物活性最好[27-28]。
为得到可能具有趋骨性的非甾体抗炎药,刘鹏等设计合成了保护的天冬氨酸六肽,并分别使用混合酸酐法和二环己基碳二亚胺(DCC)法连接布洛芬,氢化去保护基,得到L-天冬氨酸六肽-布洛芬[29]。
程利等报道了以乙二胺和缩二乙二醇为桥,采用DCC法通过酰胺键和酯键将布洛芬连接到树枝状核心上,合成得末端含2~3个布洛芬官能基的新型第1代树枝状化合物,可精确调节载药量[30]。
3 手性布洛芬及其衍生物的合成
布洛芬自上市以来,一直以外消旋形式销售。但研究表明,外消旋布洛芬的2种异构体S-布洛芬和R-布洛芬在药动学和生物转化方面不同,S-布洛芬具有明显较高的临床效果。与外消旋化合物相比,S-布洛芬可以快速在血液中达到治疗浓度。
3.1 化学拆分法
Hardy等报道以手性α-甲基苄胺(即手性苯乙胺)为拆分剂,经与消旋布洛芬成盐结晶、第1次重结晶、第2次重结晶、分解、结晶等步骤制得(S)-布洛芬,同时还报道了(S)-布洛芬-S-赖氨酸盐和钠盐的制备。成盐结晶以甲苯、甲醇为溶剂,析出产品的ee达89%,第1次重结晶后提高至94%,第2次重结晶后达98.5%。分解放出的S-布洛芬ee为98.5%,精制后ee为99%以上[31]。
单自兴报道了以氯霉素生产中的手性 “废料”L-(+)-2-氨基-1-对硝基苯基-1,3-丙二醇在三乙胺等存在下烷基化所得的L-(+)-2-N,N-二烷氨基-1-对硝基苯基-1,3-丙二醇为拆分剂,经与布洛芬成盐、纯化、析解得S-布洛芬,收率高、成本低、光学纯度(ee)为95%~99%[32]。
3.2 生物拆分法
谭天伟等报道利用衍生自亚罗解脂酵母(Yarrowia Lipolytica)的细胞外脂肪酶将外消旋布洛芬与醇一起酯化,获得S-布洛芬酯,将其水解即得S-布洛芬。所述脂肪酶可以游离的粗酶形式使用,也可以固定化酶和修饰酶等的形式使用[33]。
Giacomini等报道用还原酶将消旋2-(4-异丁基苯基)丙醛还原为手性的2-(4-异丁基苯基)丙醇,再经氧化制得手性布洛芬[34]。但该法还原时需要添加辅酶NADH,而且由手性醇氧化制S-布洛芬较难,工业化意义不大。
由于拆分时会产生不需要的R-布洛芬,因此这部分的外消旋化具有重要的意义。Young等报道了将手性2-芳基丙酸与氢氧化钠、水、甲苯混合加热,加入(S)-甲基苄胺继续加热搅拌数小时,分层,水相经脱水得消旋2-芳基丙酸[35]。
3.3 右旋布洛芬衍生物
朱志宏等报道了右旋布洛芬愈创木酚酯的制备,右旋布洛芬经SOCl2的酰氯化后与愈创木酚在吡啶/甲苯中反应得白色针状结晶产物[36]。
4 结论与展望
1,2-转位重排法因其收率高、安全性好、重排催化剂价廉易得等优点,为国内多数厂家所采用,但工艺上还有许多不足,如酰化时以石油醚作溶剂,损耗大、副产物多、安全性不高、缩酮时间长、转化率偏低,重排时副产的3-氯-2,2-二甲基-1-丙醇未得到回收或充分回收利用等。应加强对每一步反应的进一步优化,深挖潜力,提高产品的竞争力。
右旋布洛芬由于起效快,在解热镇痛作用上有显著的优点,预计今后用量比布洛芬增长快,国内需要进行重点研究,开发上市。布洛芬已经30多年临床使用,疗效确切,为最经典的非甾体消炎镇痛药之一,具有无可替代的临床地位,也是少数规模超万吨的药物之一,但同大多数非甾体消炎镇痛药一样,布洛芬也有一定的胃肠道刺激副作用,因此也必需大力加强布洛芬衍生物和制剂的研究,以提高疗效,减小副作用,提高靶向性。国外有很多成功的实例,如布洛芬吡甲酯、布洛芬精氨酸盐等。
[1]于凤丽,赵玉亮,金子林.布洛芬合成绿色化进展[J].有机化学,2003,11(11):1 198-1 204.
[2]郭莉娜,侯仲轲,陈灿,等.不对称催化反应合成手性药物的研究进展[J].精细化工中间体,2006,36(2):1-4,10.
[3]李兴泰,隋静,杨贤梅.1-氯乙基-4-异丁苯酮的绿色合成工艺:中国,1807383[P].2006-07-26.
[4]石文平,卢旭军,李国文.SO42-/Fe2O3-ZrO2-La2O3固体超强酸催化剂及其催化合成缩酮[J].精细石油化工进展,2003,4(10):33-35,85.
[5]舒瑞友.布洛芬产品中重排工艺改进[J].齐鲁药事,2006,25(4):242-243.
[6]Manimaran T,Harkins A E.Process for producing high purity ketones by Friedel-crafts acylation at low temperature:WO,2007044270[P].2007-04-19.
[7]Chaudhari R V,Majeed S A,Seayad J.Process for the preparation of ibuprofen:US,6093847[P].2000-07-25.
[8]Wu T C.Preparation of aralkanoic acids and esters using mixed ligand catalyst:US 5902898[P].1999-05-11.
[9]Elango V.Process for the carbonylation of arylalkyl halides:US,6555704[P].2003-04-29.
[10]Chan A S C,Pai C C.Chiral 4,4'-bis(disubstitutedphosphino)-3,3'-bipyridines,their use in catalysts for asymmetric hydrogenation of 2-arylpropenoic acids and preparation thereof:GB,2332201[P].1999-06-16.
[11]姜标,司玉贵,陈君,等.一种制备2-芳基乳酸酯及萘普生、布洛芬的方法:中国,1706802[P].2005-12-14.
[12]陈平,刘春华,黄鹏勉,等.布洛芬的制备方法:中国,101456808[P].2009-06-17.
[13]Akbarali P M,Vijayaraj K K,Gani R S,et al.Novel process for producing ibuprofen sodium dihydrate:WO,2005121061[P].2005-12-22.
[14]董雪伶,徐砚珂,李振志.L-精氨洛芬的制备[J].中国医药工业杂志,2002,33(2):58-59.
[15]刘杏敏.一种布洛芬精氨酸盐的制备方法:中国,101239901[P].2008-08-13.
[16]王润玲.一种精氨酸布洛芬盐的制备方法:中国,101190889[P].2008-06-04.
[17]周金森,廖成竹,冯小龙,等.布洛芬胺盐的合成与结构表征[J].分析测试学报,2004,23(4):18-21.
[18]宋妮,李英霞,孙雪.一种布洛芬季铵盐衍生物的合成[J].青岛海洋大学学报,2002,32(6):911-913.
[19]刘勇,罗氚芸,李蕾,等.布洛芬乙酯的分子筛催化合成[J].化学世界,2000,(2):90-92.
[20]向玲,吴贝,邓勇.酰氯法合成布洛芬吡甲酯工艺的改进[J].华西药学杂志,2006,21(2):178-179.
[21]王鹏.布洛芬酯、可药用盐及其制备工艺以及它的药物组合物:中国,1640870[P].2005-07-20.
[22]孙礼林,孙玉,汪凌云,等.布洛芬高分子前体药物及纳米微球的合成和表征[J].功能高分子学报,2004,17(1):97-101.
[23]赵秀丽,陈大为,李可欣,等.布洛芬丁香酚酯前体药物的合成及其水解动力学[J].沈阳药科大学学报,2006,23(2):70-73.
[24]赵一玫,夏丹,艾彩萍,等.布洛芬衍生物的合成及抗炎活性[J].中国药物化学杂志,2005,15(6):360-362,366.
[25]魏东芝,赵相国,宋庆训.一种布洛芬糖衍生物及其制备方法和应用:中国,1683385[P].2005-10-19.
[26]王化录.布洛芬三氮唑核苷酯及制备方法和用途:中国,1379037[P].2002-11-13.
[27]宋妮,李英霞,孙雪,等.布洛芬糖衍生物的合成[J].药学学报,2004,39(2):105-109.
[28]宋妮,李英霞,李春霞,等.一种布洛芬糖缀合物及其制备方法和应用:中国,1513861[P].2004-07-21.
[29]刘鹏,张昱,郭丽.L-天冬氨酸六肽-布洛芬的合成[J].华西药学杂志,2007,22(1):49-50.
[30]程利,彭莉,郭丽.第一代新型布洛芬树枝状化合物的合成[J].华西药学杂志,2009,24(2):112-114.
[31]Hardy R,Coe P F,Hirst A,et al.Process of resolving phenylpropionicacidsusingα-methylbenzylamine:US,5599969[P].1997-02-04.
[32]单自兴.一种(S)-布洛芬的制备方法:中国,1126729[P].2003-11-05.
[33]谭天伟,刘英,王芳.生物催化制备S-布洛芬及S-布洛芬酯的方法:中国,101104861[P].2008-01-16.
[34]Giacomini D,Cainelli G,Galletti P,et al.Process for the preparationofchiral2-arylpropylicalcohols:WO,2008074717[P].2008-06-26.
[35]Young R E,Phan H V,Manimaran T,et al.Racemization process for optically active carboxylic acids or esters thereof:US,5847202,1998-12-08.
[36]朱志宏,郑良彬,卜振军.右旋布洛芬愈创木酚酯及其制备方法:中国,101456814[P].2009-06-17.
Advances on the Syntheses of Ibuprofen,Dexibuprofen and Derivatives
Wang Jiahua1,Zheng Tucai2,Wang ping3
(1.Juhua Group Pharmaceutical Factory,Quzhou,Zhejiang 324000;2.Department of Chemical and Pharmaceutical Engineering,West Branch of Zhejiang University of Technology,Quzhou,Zhejiang 324000;3.College of pharmaceutical Scjences,Zhejiang University of Technology,Hang Zhou 310014)
This paper first reviews and comments on six synthetic routes of ibuprofen,namely,migration rearrangement,alcohol carbonylation,alkene carbonylation,alkyl halide carbonylation,alkene catalytic hydrogenation and oxirane rearrangement.Then it summarizes the syntheses and characteristics of ibuprofen derivatives such as l-arginine salt,various esters and amides.The preparation of dexibuprofen and derivatives is then briefly discussed.Migration rearrangement is still the best method for preparing ibuprofen,and the key technical points are emphasized,together with the hope for further research on dexibuprofen and ibuprofen derivatives.
ibuprofen;dexibuprofen;carbonylation;migration rearrangement;resolution;synthesis
TQ 463+.25
ADOI10.3969/j.issn.1006-6829.2010.02.008
2009-01-09;
2010-02-02
