全乙酰GM3三糖对甲氧基苯酚苷的合成*
2010-11-26李相鹏李英霞
李相鹏, 王 鹏, 李英霞
(1. 中国海洋大学 医药学院,山东 青岛 266003; 2. 复旦大学 药学院,上海 201203)
神经节苷脂GM3(Neu5NAcα2→3Galβ1→4Glc1→1Cer)于1952年由Yamakawa等[1]从马血红细胞中首次分离得到,其亲酯性神经酰胺基团包埋于细胞膜磷酸双分子脂层中,而亲水性糖链部分伸向细胞膜表面。GM3在细胞膜的信号转导中起到了重要作用,对细胞的粘附、分化、增殖与癌变等过程起到了调节作用[2],其化学合成工作已经成为目前研究的热点[3~14]。
本研究小组[14]在以往的研究过程中,以唾液酸N-苯基三氟亚胺酯(10)为供体,6位TBDPS(叔丁基二苯基硅基)保护的半乳糖衍生物为受体,在温和促进剂TBSOTf(叔丁基二甲基硅三氟甲磺酸酯)的条件下,高效地构建了α-2,3糖苷键,并将此策略成功的应用于GM3的合成。该方法采用线性合成策略,唾液酸(7)需要依次连接半乳糖、葡萄糖和鞘氨醇,所需步骤较长,产物不易积累。
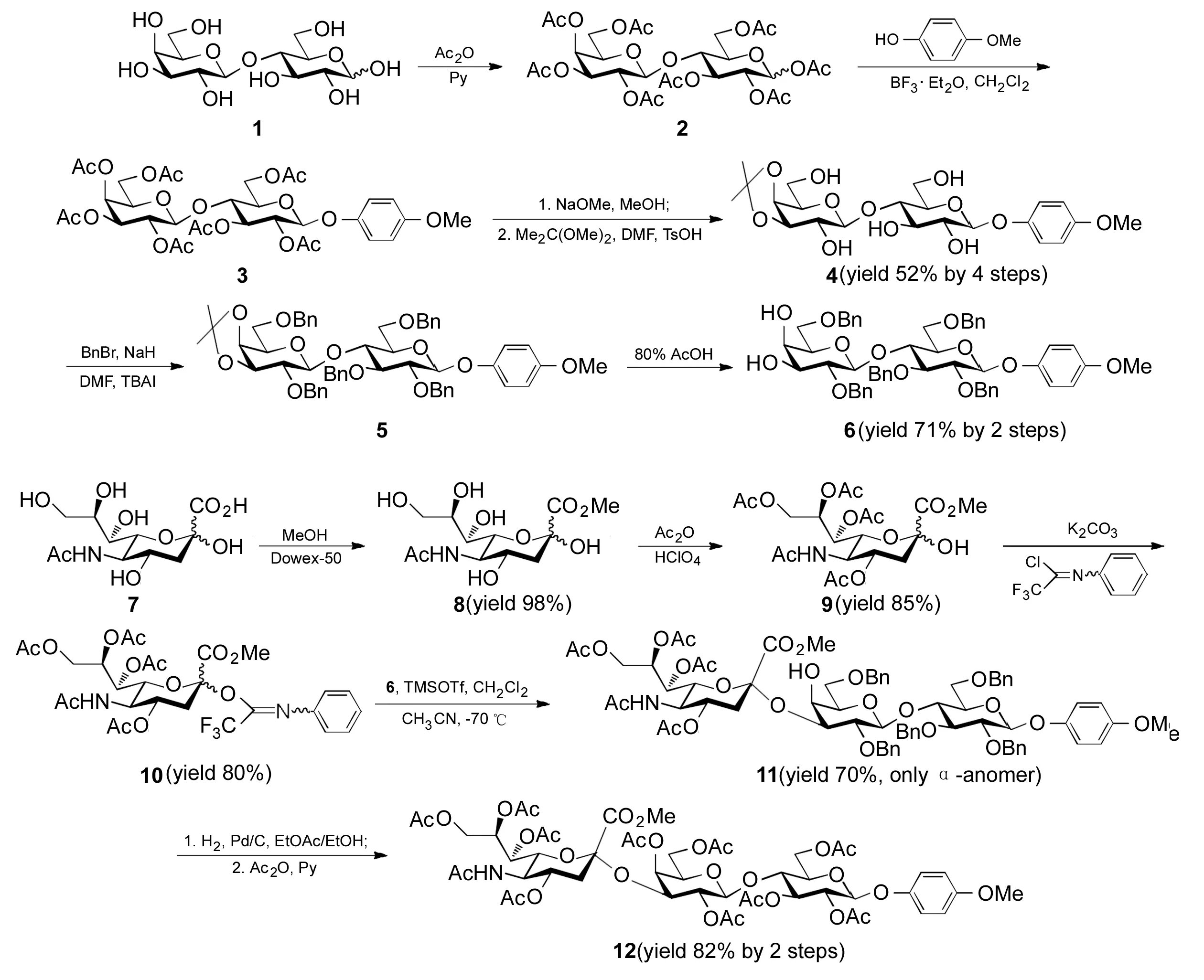
Scheme 1
本文分别以乳糖(1)和7为原料,制得受体乳糖二醇(6)和供体10;6与10经糖苷化以较高收率(70%)制得α-构型的神经节苷脂GM3三糖衍生物(11); 将11的保护基Bn转化为Ac制得GM3全合成的关键中间体——全乙酰GM3三糖对甲氧基苯酚苷(12, Scheme 1),其结构经1H NMR,13C NMR和HR-MS确证。
1 实验部分
1.1 仪器与试剂
JEOL JNM-ECP 600 MHz型核磁共振波谱仪(DMSO-d6为溶剂,TMS为内标); Q-TOF Global型质谱仪(ESI)。
TMSOTf和Dowex-50, Aldrich-Sigma-Acros;其余所用试剂均为国产分析纯;CH2Cl2经CaH回流重蒸处理,CH3CN经P2O5回流重蒸;DMF,吡啶、甲醇经分子筛干燥处理。所有反应如非特殊说明均在氩气保护下进行。
1.2 合成
(1) 4的合成
在圆底瓶中加入1 5.0 g(14.6 mmol)和吡啶100 mL,搅拌形成混悬液,冰浴冷却,滴加醋酐16.5 mL(175 mmol),滴毕,于室温反应过夜。减压浓缩后加入CH2Cl2300 mL,依次用稀盐酸(3×100 mL),饱和NaHCO3溶液(3×100 mL),饱和NaCl溶液(3×100 mL)洗涤,无水Na2SO4干燥,减压浓缩得淡黄色黏稠物(2)。用CH2Cl2(300 mL)溶解,加入对羟基苯甲醚1.8 g(14.6 mmol),冰浴冷却,搅拌下滴加三氟化硼乙醚2.2 mL(24.8 mmol),滴毕,自然恢复至室温反应2 h。用三乙胺调至中性,减压浓缩,残余物经硅胶柱层析[洗脱剂:A=V(乙酸乙酯) ∶V(石油醚)=1 ∶3]分离得无色浆状物(3)。用CH2Cl2(50 mL)溶解,加入甲醇50 mL和甲醇钠780 mg(1.46 mmol),搅拌下于室温反应2 h(析出大量白色固体)。加入酸性树脂调至中性,倾出上层混悬液减压浓缩得白色固体。将其混悬于DMF(20 mL)中,加入二甲氧基丙烷20 mL和对甲苯磺酸27 mg(0.15 mmol),反应2 h。减压浓缩,残余物经硅胶柱层析[洗脱剂:V(乙酸乙酯) ∶V(MeOH)=25 ∶1]分离得白色固体4 3.7 g,产率52%(以1计算);1H NMRδ: 6.99~6.97(m, 2H, ArH), 6.87~6.84(m, 2H, ArH), 5.48(d,J=5.5 Hz, 1H, 2a-OH), 4.80(d,J=7.7 Hz, 1H, 1a-H), 4.68~4.66(m, 2H, 3b,6a-OH), 4.29(d,J=8.2 Hz, 1H, 1b-H), 4.11(dd,J=5.5 Hz, 1.8 Hz, 1H, 4b-H), 3.97(dd,J=7.4, 6.0 Hz, 1H, 3b-H), 3.85(ddd,J=7.3 Hz, 5.0 Hz, 1.8 Hz, 1H, 5b-H), 3.74~3.71(m, 1H, 6a-H), 3.70(s, 3H, OCH3), 3.64~3.52(m, 3H, 6a,6b-H), 3.47~3.45(m, 1H, 5a-H), 3.44~3.40(m, 2H, 3a,4a-H), 3.28~3.23(m, 2H, 2a,2b-H), 1.40(s, 3H, CH3), 1.25(s, 3H, CH3)。
(2) 6的合成
冰浴冷却,在圆底瓶中加入4 1.00 g(2.05 mmol)的DMF(50 mL)溶液和NaH 370 mg(15 mmol),搅拌下于室温反应30 min;加入TBAI(四丁基碘化铵)20 mg,冰浴冷却下滴加BnBr(溴苄)1.46 mL(12.2 mmol),滴毕,于室温反应2 h。冰浴冷却下用甲醇终止反应,减压浓缩,残余物用CH2Cl2(200 mL)稀释,依次用稀盐酸(3×80 mL), 饱和NaHCO3溶液(3×80 mL),饱和NaCl溶液(3×80 mL)洗涤,无水Na2SO4干燥,减压浓缩得微黄色浆状物(5)。加入80%AcOH(50 mL),于80℃反应2 h,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:A=1 ∶1)分离得白色无定形固体61.31 g,产率71%(以4计算);1H NMRδ: 7.42~7.40(m, 4H, AHr), 7.32~7.24(m, 21H, ArH), 7.01~7.99(m, 2H, ArH), 6.84~6.81(m, 2H, ArH), 5.07(d,J=7.3 Hz, 1H, 1a-H), 5.04(d,J=10.5 Hz, 1H, PhCH2), 4.99(br s, 1H, OH), 4.86(d,J=11.5 Hz, 1H, PhCH2), 4.79~4.72(m, 4H, OH, PhCH2), 4.63(d,J=11.0 Hz, 1H, PhCH2), 4.48(d,J=11.9 Hz, 1H, PhCH2), 4.41(d,J=7.1 Hz, 1H, 1b-H), 4.39(d,J=11.5 Hz, 1H, PhCH2), 4.34(d,J=11.9 Hz, 1H, PhCH2), 4.31(d,J=11.9 Hz, 1H, PhCH2), 3.86~3.80(m, 2H, 3a,6a-H), 3.78~3.72(m, 2H, 4a,5a-H), 3.70(s, 3H, OCH3), 3.69~3.67(m, 2H, 6a,4b-H), 3.63(dd,J=9.6 Hz, 5.5 Hz, 1H, 6b-H), 5.31(t,J=5.9 Hz, 1H, 5b-H), 3.47~3.40(m, 4H, 2a,2b,3b,6b-H) 。
(3)8的合成
在反应瓶中加入71.5 g(4.8 mmol)与Dowex-50 4 g的无水甲醇(105 mL)溶液,搅拌下于室温反应2 h。抽滤,滤饼用无水甲醇洗涤,合并滤液与洗液,浓缩干燥得白色固体81.53 g,产率98%;1H NMR(CD3OD)δ: 4.05~4.00(m, 1H, 8-H), 3.98(dd,J=10.6 Hz, 1.5 Hz, 1H, 7-H), 3.82~3.77(m, 2H, 4,5-H), 3.77(s, 3H, OCH3), 3.71~3.68(m, 1H, 9-H), 3.61(dd,J=11.3 Hz, 5.8 Hz, 1H, 9-H), 3.47(dd,J=9.1 Hz, 1.4 Hz, 1H, 6-H), 2.20(dd,J=12.8 Hz, 4.8 Hz, 1H, 3eq-H), 2.00(s, 3H, NCOCH3), 1.88(dd,J=12.8 Hz, 11.7 Hz, 1H, 3ax-H)。
(4)9的合成
在反应瓶中于40 ℃加入Ac2O 0.33 mL和60%HClO42.5μL,搅拌均匀后分批加入8110 mg(0.34 mmol)(30 min内),反应2 h。冷却至室温,加入冷水(5 mL)淬灭反应,加入NH4Cl至饱和,用CH2Cl2(3×20 mL)萃取,合并萃取液,依次用饱和NaHCO3溶液(5 mL),饱和NaCl溶液(5 mL)洗涤,无水Na2SO4干燥,浓缩后经硅胶柱层析(洗脱剂:A=3 ∶1)分离得微黄色固体9148 mg,产率89%;1H NMR(CDCl3)δ: 5.94(m, 1H, NH), 5.38(dd,J=5.5 Hz, 2.3 Hz, 1H, 7-H), 5.25~5.26(m, 1H, 8-H), 5.18~5.24(m, 1H, 4-H), 4.82(m, 1H, OH), 4.56(dd,J=12.4 Hz, 1H, 5-H), 4.23(dd,J=10.6 Hz, 1.9 Hz, 1H, 6-H), 4.17(q,J=10.1 Hz, 1H, 9-H), 4.03(dd,J=12.4 Hz, 7.8 Hz, 1H, 9-H), 3.86(s, 3H, OCH3), 2.19~2.27(m, 2H, 3eq,3ax-H), 2.15, 2.11, 2.03, 2.01, 1.90(5s, 15H, CO2CH3)。
(5)10的合成
于0 ℃在反应瓶中加入PPh351.8 g(200 mmol)和三乙胺11 mL(80 mmol)的CCl4(32 mL)悬混液,三氟乙酸5 mL(65 mmol),搅拌10 min后加入苯胺7.3 mL(80 mmol)的CCl4(31 mL)溶液,回流反应4 h。冷却至室温,倾出上清液,残余固体用正己烷洗涤数次,合并上清液及洗涤溶液,减压蒸除溶剂,残余物减压蒸馏得浅黄色液体N-苯基三氟亚胺氯。
在反应瓶中加入9 492 mg(1 mmol)的丙酮(20 mL)溶液,K2CO3414 mg(3 mmol)和N-苯基三氟乙酰氯3.1 g(15 mmol),搅拌下于室温反应2 h。过滤,滤液加压浓缩,残余物经硅胶柱层析(洗脱剂:A=3 ∶2)分离得微黄色固体10599 mg,产率85%(α∶β=1 ∶1);1H NMR(CDCl3)δ:β-anomer: 6.72~7.29(m, 5 H, ArH), 5.49(d,J=9.9 Hz, 1H, NH), 5.44(dd,J=4.4 Hz, 2.2 Hz, 1H, 7-H), 5.26 (td,J=11.1 Hz, 4.9 Hz, 1H, 4-H), 5.13~5.14(m, 1H, 8-H), 4.57(dd,J=12.7 Hz, 2.2 Hz, 1H, 9a-H), 4.28(q,J=10.4 Hz, 1H, 5-H), 4.21(dd,J=10.4 Hz, 2.2 Hz, 1H, 6-H), 4.08(dd,J=12.7 Hz, 7.7 Hz, 1H, 9b-H), 3.83(s, 3H, CO2CH3), 2.81(dd,J=13.8 Hz, 5.0 Hz, 1H, 3eq-H), 2.18(m, 1H, 3ax-H), 2.16, 2.09, 2.06, 1.91, 1.77(5s, 15H, CO2CH3, NCOCH3);α-anomer: 6.72, 7.60(m, 5H, ArH), 5.57(d,J=9.9 Hz, 1H, NH), 5.36(dd,J=6.2 Hz, 1.8 Hz, 1H, 7-H), 5.24(dd,J=6.2 Hz, 1.8 Hz, 1H, 8-H), 5.12(dt,J=10.3 Hz, 4.7 Hz, 1H, 4-H), 4.68(dd,J=10.6 Hz, 1.8 Hz, 1H, 6-H), 4.38(dd,J=12.4 Hz, 2.9 Hz, 1H, 9a-H), 4.24(dd,J=12.4 Hz, 6.3 Hz, 1H, 9b-H), 4.13~4.18(m, 1H, 5-H), 3.83(s, 3H, CO2CH3), 2.74(dd,J=13.6 Hz, 5.1 Hz, 1H, 3eq-H), 2.32(dd,J=13.6 Hz, 10.6 Hz, 1H, 3ax-H), 2.17, 2.04, 1.99, 1.97, 1.92(5s, 15H, CO2CH3, NCOCH3)。
(6)11的合成
在两口瓶中加入10500 mg(0.75 mmol),6452 mg(0.50 mmol), CH2Cl225 mL, CH3CN 25 mL和4 Å分子筛1 g,于室温搅拌30 min后将两口瓶置于-70 ℃低温反应装置中,10 min后以微量进样器加入TMSOTf 18μL(0.10 mmol), 于-70 ℃反应1.5 h。 用三乙胺终止反应,逐渐升至室温后抽滤除去分子筛,滤液浓缩后经硅胶柱层析[洗脱剂:B=V(CH2Cl2) ∶V(MeOH)=100 ∶1]分离得白色无定型固体11480 mg,产率70%;1H NMR(CDCl3)δ: 7.42~7.41(m, 2H, ArH), 7.34~7.22(m, 23H, PhH), 7.02~7.01(m, 2H, PhH), 6.78~6.77(m, 2H, PhH), 5.42(ddd,J=8.2 Hz, 5.9 Hz, 2.3 Hz, 1H, 8c-H), 5.32(dd,J=8.2 Hz, 2.3 Hz, 1H, 7c-H), 5.15(d,J=10.1 Hz, 1H, NH), 5.01(d,J=11.0 Hz, 1H, PhCH2), 4.98(d,J=11.0 Hz, 1H, PhCH2), 4.88~4.85(m, 1H, 4c-H), 4.84(d,J=7.4 Hz, 1H, 1a-H), 4.81(d,J=10.1 Hz, 1H, PhH), 4.80(d,J=11.9 Hz, 1H, PhCH2), 4.77(d,J=10.5 Hz, 1H, PhCH2), 4.69(d,J=11.9 Hz, 1H, PhCH2), 4.58(d,J=7.8 Hz, 1H, 1b-H), 4.43(d,J=11.9 Hz, 1H, PhCH2), 4.34(d,J=11.9 Hz, 1H, PhCH2), 4.31(dd,J=12.4 Hz, 2.8 Hz, 1H, 9a-H), 4.10(d,J=11.0 Hz, 1H, PhCH2), 4.07(d,J=11.0 Hz, 1H, PhCH2), 3.96(dd,J=12.4 Hz, 5.9 Hz, 1H, 9a-H), 3.77(s, 6H, OCH3), 3.56(dd,J=9.1 Hz, 7.7 Hz, 1H, 2b-H), 2.51(dd,J=13.3 Hz, 4.6 Hz, 1H, 3c eq-H), 2.10(s, 3H, COCH3), 2.07(t like,J=12.4 Hz, 12.0 Hz, 1H, 3c ax-H), 2.02(s, 3H, COCH3), 2.00(s, 3H, COCH3), 1.89(s, 3H, COCH3), 1.88(s, 3H, COCH3);13C NMR(CDCl3)δ: 170.8, 170.6, 170.3, 170.0, 169.9, 168.3, 155.1, 151.6, 139.0, 138.9, 138.5, 138.3, 128.3, 128.3, 128.2, 128.1, 128.1, 128.0, 127.6, 127.5, 127.5, 127.4, 127.3, 127.2, 118.4, 114.4, 102.7, 102.5, 98.4, 83.0, 81.6, 78.4, 77.4, 76.6, 76.3, 75.5, 75.2, 75.1, 74.9, 69.0, 68.7, 68.4, 67.9, 67.1, 62.2, 55.6, 49.2, 36.5, 29.7, 23.2, 21.2, 20.7, 20.5; HR-MS: Calcd for C74H85NO24Na{[M+Na]+} 1 394.535 9, found 1 394.539 1。
(7)12的合成
在单口瓶中加入11300 mg(0.21 mmol)的乙酸乙酯(20 mL)-乙醇(20 mL)溶液和Pt/C 100 mg,接上回流管及带有氢气球的三通阀,搅拌下回流反应10 h。抽滤除去Pt/C,滤液浓缩得浆状物,用吡啶(10 mL)溶解后再加入醋酐3 mL,于室温反应8 h。减压浓缩,用CH2Cl2(30 mL)稀释,依次用稀盐酸(3×10 mL),饱和NaHCO3溶液(3×10 mL),饱和NaCl溶液(3×10 mL)洗涤,无水Na2SO4干燥,减压浓缩后经硅胶柱层析(洗脱剂:B=50 ∶1)分离得白色无定型固体12207 mg,产率82%;1H NMR(CDCl3)δ: 6.93~6.91(m, 2H, PhH), 6.81~6.79(m, 2H, PhH), 5.55(ddd,J=9.6 Hz, 5.0 Hz, 2.3 Hz, 1H, 8c-H), 5.38(dd,J=9.2 Hz, 2.8 Hz, 1H, 7c-H), 5.25(t like,J=9.2 Hz, 9.1 Hz, 1H, 3a-H), 5.15~5.12(m, 2H, NH, 2a-H), 4.94(dd,J=8.2 Hz, 7.4 Hz, 1H, 2b-H), 4.91(d,J=7.8 Hz, 1H, 1a-H), 4.89~4.86(m, 2H, 4,4b-H), 4.70(d,J=7.8 Hz, 1H, 1b-H), 4.52(dd,J=10.1 Hz, 3.2 Hz, 1H, 3b-H), 4.47(dd,J=11.9 Hz, 2.3 Hz, 1H, 6a-H), 4.41(dd,J=12.4 Hz, 2.8 Hz, 1H, 9c-H), 4.21(dd,J=11.9 Hz, 5.9 Hz, 1H, 6a-H), 4.06~3.95(m, 5H, 6a,4b,5b,5c,9c-H), 3.85(m, 1H, 6b-H), 3.84(s, 3H, OCH3), 3.76(s, 3H, OCH3), 3.73~3.71(m, 1H, 5a-H), 3.63(dd,J=11.0 Hz, 2.3 Hz, 1H, 6c-H), 2.57(dd,J=12.4 Hz, 4.6 Hz, 1H, 3c eq-H), 2.25(s, 3H, COCH3), 2.16(s, 3H, COCH3), 2.08~2.06(m, 21H, COCH3), 2.00(s, 3H, COCH3), 1.85(s, 3H, NCOCH3), 1.68(t,J=12.4 Hz, 1H, 3cax-H);13C NMR(CDCl3)δ: 170.9, 170.7, 170.6, 170.4, 170.3, 170.2, 169.7, 169.6, 169.6, 167.9, 155.6, 150.9, 118.6, 114.5, 101.0, 99.9, 96.7, 76.3, 73.4, 72.8, 71.6, 71.3, 70.5, 69.3, 76.7, 67.3, 66.9, 62.2, 61.5, 55.6, 53.1, 49.1, 41.1, 37.3, 29.7, 23.1, 21.5, 20.9, 20.8, 20.7, 20.7, 20.6; HR-MS: Calcd for C51H67NO30Na{[M+Na]+} 1 196.364 6; found 1 196.366 2。
2 结果与讨论
2.1 乳糖受体的设计
基于简单易得并可大量制备的考虑,设计了受体6,对甲氧基苯酚苷作为异头碳保护基,可在较温和的条件下脱除并转化为三糖供体与鞘氨醇偶联[13],而苄基保护除3,4-位以外的羟基,虽然不具有邻基参与作用,但其可以方便的脱除转化为酰基,同时苄基的供电子效应可以提高受体亲核性。
2.2 糖苷化反应催化剂的选择
11的合成首先采用了以往的糖苷化条件[14],以 TBSOTf为催化剂,反应过程非常缓慢,即使将反应温度提高到室温也只有少量供体分解,于室温反应24 h,产率仅为40%。改用酸性较强的TMSOTf为催化剂,反应在1.5 h内结束,且收率提高至70%。其原因可能是TBSOTf催化活性较弱,反应以SN1历程为主,而乳糖受体较大位阻使得亲核进攻难以发生;而高活性催化剂TMSOTf可使供体分解为氧鎓离子中间体,可促进反应。
2.3 11和12糖苷键构型的确定
11和12的NMR数据与文献报道[13]一致,确定为α-构型。以TBSOTf或MSOTf为催化剂,产物均为α-构型。推测原因为乙腈的溶剂效应及乳糖受体6的特殊空间结构使得难以生成β-构型产物。
总之,本文首次以唾液酸N-苯基三氟亚胺酯10为供体与乳糖受体6进行了糖苷化尝试,以极高的产率、立体选择性得到了GM3三糖衍生物,并将其保护基全部转化为乙酰基,得到了GM3全合成的关键中间体12。这将使GM3及其衍生物的合成更加简洁有效,为其生物活性的深入研究解决了材料来源问题。
[1] Yamakawa T, Suzuki S. The chemistry of the lipids of posthemolytic residue or stroma of erythrocytes.Ⅲ.Globoside,the sugar-containing lipid of human blood stroma[J].J Biochem,1952,39(4):393-402.
[2] Prokazova N V, Samovilova N N, Gracheva E V,etal. Ganglioside GM3and its biological functions[J].Biochemistry (Moscow),2009,74(3):235-249.
[3] Numata M, Sugimoto M, Shibayama S,etal. A total synthesis of hematoside,αNeuGc(2-3)-βGal(1-4)-βGlc(1-1)Cer[J].Carbohydr Res,1988,174(1):73-85.
[4] Murase T, Ishida H, Kiso M,etal. A facile,regio- and stereo-selective synthesis of ganglioside GM3[J].Carbohydr Res,1989,188(1):71-80.
[5] Hasegawa A, Murase T, Morita M,etal. Synthetic studies on sialoglycoconJugates 15:Synthesis of ganglioside GM3analogs containing a variety of lipophilic parts[J].J Carbohydr Chem,1990,9(2-3):201-214.
[6] Ito Y, Paulson J C. A novel strategy for synthesis of ganglioside GM3using an enzymically produced sialoside glycosyl donor[J].J Am Chem Soc,1993,115(4):1603-1605.
[7] Liu K K C, Danishefsky S. A striking example of the interfacing of glycal chemistry with enzymatically mediated sialylation:A concise synthesis of ganglioside GM3[J].J Am Chem Soc,1993,115(11):4933-4934.
[8] Tomoo T, Kondo T, Abe H,etal. An efficient short-step total synthesis of ganglioside GM3:Effective usage of the neighbouring group participation strategy[J].Carbohydr Res,1996,284(2):207-222.
[9] Zehavi U, Tuchinsky A. Enzymic glycosphingolipid synthesis on polymer supports.Ⅲ.Synthesis of GM3,its analog [NeuNAcα(2-3)Galβ(1-4)Glcβ(1-3) Cer] and their lyso-derivatives[J].GlycoconJugate J,1998,15(7):657-662.
[10] Lutz F T, Dirk G. Synthesis of Specifically Labelled Ganglioside[1c-13C]-GM3[J].Eur J Org Chem,1998,9:1895-1899.
[11] Mauri L, Casellato R, Kirschner G,etal. A procedure for the preparation of GM3ganglioside from GM1-lactone[J].GlycoconJugate J,1999,16(3):197-203.
[12] Richard I, Duclos Jr. The total synthesis of ganglioside GM3[J].Carbohydr Res,2000,328(4):489-507.
[13] 朱振元,张勇民. 神经节苷脂GM3的有效合成[J].化学学报,2007,65(24):2909-2916.
[14] Liu Y, Ruan X, Li X,etal. Efficient synthesis of a sialic acidα(2→3)galactose building block and its application to the synthesis of ganglioside GM3[J].J Org Chem,2008,73(11):4287-4290.
