水热法合成纳米MoS2研究与应用进展
2024-01-31郭维敏李树昌闫发发张振威郑顺奇
郭维敏,李树昌,闫发发,张振威,郑顺奇
(中国兵器科学研究院宁波分院,浙江 宁波 315000)
1 前 言
从世界范围来看,我国的钼资源储量较为丰富。根据国土资源部2017年发布的数据,截止2016年末,我国钼矿查明资源储量为2882.41万吨(金属含量)[1]。因此,围绕钼元素开展科学研究意义重大。
钼元素位于元素周期表第ⅥB族、第5周期,为过渡金属元素。金属钼是合金钢中一种重要的成分,能够提高钢的淬透性,增加回火稳定性。钼原子的价电子轨道呈半充满状态,表现为典型的过渡状态。当钼原子与硫原子化合后,就形成了一系列化合物,其中二硫化钼(MoS2)最为常见。MoS2具有典型的层状结构,单层的MoS2由上、下两层硫离子组成的六角平面中间夹一层钼离子构成。MoS2主要有3种堆垛方式,分别是1T型、2H型和3R型,它们的结构如图1[2]所示。在2H-MoS2和3R-MoS2中每个钼离子位于硫离子构成的正三棱柱的中心。层内钼离子与硫离子以较强的离子键结合,层间以微弱的范德华力相作用。2H-MoS2和3R-MoS2的物理性质极为相似,是辉钼矿的主要成分。1T-MoS2是一种亚稳相,在高温退火后会转化为2H-MoS2。
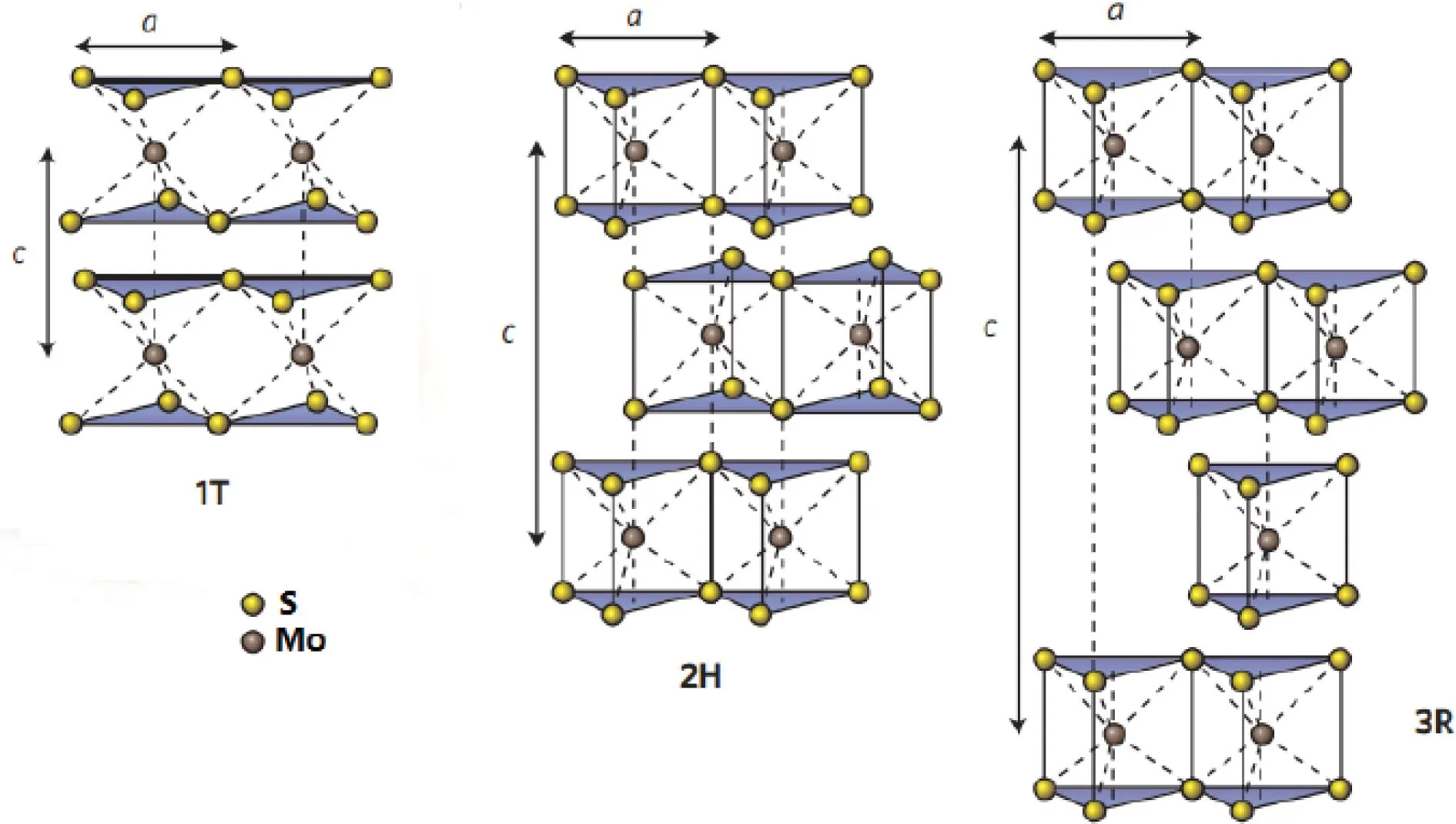
图1 1T、2H、3R 构型的MoS2的晶体结构[2]Fig.1 Crystal structure of 1T-MoS2,2H-MoS2 and 3R-MoS2[2]
正因为这种特殊的层状结构,MoS2广泛应用于润滑[3]和催化[4]等领域。MoS2层与层之间的作用力很小,当受到很小的剪切力时,层与层之间发生滑动;它对金属具有很强的吸附能力,在高温、高速、高压、高真空或低温等极端恶劣工作条件下能够保持良好的润滑效果,延长机械的寿命。在催化领域,MoS2的催化活性往往与它暴露的活性位点有关[5]。这些活性位点位于MoS2层状结构的边缘,通过合成一些特殊形貌结构,如纳米线、多孔、树突等可以增加活性位点的浓度。在水热合成中,可以通过控制合成条件在层状结构的底面上形成空洞缺陷产生新的边缘。水热法不仅能制备出形貌各异的纳米MoS2,还能与其他材料复合提升储能[6]和电催化性能[7]。纳米MoS2及其复合材料具有丰富的活性位点和大的比表面积,是近年来研究的热点之一。本文将重点介绍水热法合成的纳米MoS2在储能、光催化、电催化析氢领域的研究进展。
2 纳米MoS2的合成方法
纳米MoS2的合成方法可分为“自上而下”法和“自下而上”法。“自上而下”法是指以块体或者多层的MoS2为原料,通过插层、剥离等方法制备纳米MoS2,其主要的制备方法有机械剥离法[8]、超声辅助剥离法[9]和插层剥离法[10]等,是一种以物理作用为主的制备方法。“自下而上”法是以特定的钼源和硫源为原料合成纳米MoS2的一类方法,其主要方法有水热法(溶剂热法)[11,12]和化学气相沉积法[13],以化学作用为主。
2.1 插层剥离法
插层剥离法是指先通过化学反应在MoS2层间嵌入锂原子[14],再利用锂原子与水或醇反应来扩大层间距,从而获得少层或单层的纳米MoS2。Ortiz-Quiles等[14]将MoS2固体粉末放入正丁基锂的正己烷溶液中进行嵌锂,之后再将产物分离投入水中进行剥离。X射线衍射表征发现其峰向小角度偏移,衍射峰强度降低、半高宽增加,表明剥离效果较好,但SEM照片中已观察不到层状结构。在插层剂强烈的作用下,MoS2的片层结构会受到一定程度的损伤。
2.2 超声辅助剥离法
超声辅助法是指利用超声波产生的能量使溶剂分子进入块体MoS2的分子层间,块体的MoS2逐渐被剥离成片层状,收集产物获得纳米MoS2。溶剂的表面张力是影响剥离效果的重要因素。Coleman等[15]的实验与理论计算结果表明:在溶剂表面张力为40 mN·m-1左右时,离心后上清液中MoS2的浓度最高。Singh等[16]在去离子水中添加表面活性剂CTAB调节表面张力,然后加入MoS2固体粉末对其进行超声剥离。MoS2产物XRD衍射峰与拉曼光谱变化表明,添加CTAB的水溶液对MoS2固体粉末具有良好的剥离效果。虽然超声辅助剥离操作简单,获得的纳米片缺陷较少,但它的效率较低,对超声设备的性能要求较高,往往需要数小时超声处理才能获得低浓度(5 mg·mL-1左右)的MoS2纳米片分散液。
2.3 化学气相沉积法
化学气相沉积(chemical vapor deposition,CVD)法是指在一定的保护气氛下,将气态反应物送到反应装置内,在特定的装置内发生化学反应,最终在基底上生成固态沉淀物。对于MoS2,一般以三氧化钼为钼源、单质硫或硫化氢作硫源来合成。王巍等[17]以硫粉为硫源,三氧化钼为钼源,在管式炉中采用CVD法制备了纳米管状MoS2。这种方法能够获得结构完整的片层结构,但产率低,对设备的密闭性及控温精度要求较高。
2.4 水热法和溶剂热法
水热法是以密封在容器中的水为介质,在一定的温度和压力下,使常温常压不溶于水的物质溶解并发生化学反应或再结晶反应,反应产物过饱和析出的一种制备纳米材料的方法。水热法制备MoS2常用的钼源有仲钼酸铵、钼酸钠和三氧化钼,常用的硫源有单质硫、硫化钠、硫代硫酸钠和硫脲等。在合成过程中加入表面活性剂可以改变纳米颗粒不同方向的生长速率,从而改变MoS2的形貌。
早在1845年,Eschafhautl采用水热法以硅酸为原料制备出了石英晶体[18]。在早期,水热法主要用于成矿机理的研究。1948年,Barrer等[18]模拟天然沸石的形成环境合成了人造沸石。1985年,Bibby等[19]在非水溶剂中合成了沸石,溶剂热法合成纳米材料进入了人们的视野之中。溶剂热法的出现弥补了水热法的不足,能够合成易水解和对水敏感的硫化物。水热法和溶剂热法的出现为纳米材料的合成与制备带来了新方法,有效地促进了纳米材料的发展。
3 纳米MoS2的水热合成与应用
水热法是制备纳米MoS2的重要方法。通过改变水热合成原料初始浓度、反应温度和时间等条件,可以调控纳米MoS2的层间距、形貌等特性。这些特性与储锂、催化活性等性能密切相关。下面将从储能领域、光催化领域和电催化析氢领域,介绍水热法合成纳米MoS2的研究进展。
3.1 储能领域
超级电容器是一种新型的储能装置,它寿命长,充电时间短,可大电流充放电,具有较高的能量密度和功率密度[20]。Ma等[21]在水热合成2H-MoS2的过程中加入了海藻酸钠(sodium alginate,SA),制备了花状结构的MoS2球。其制备过程如图2a所示;海藻酸钠中含有丰富羟基和羧基,能够诱导形成花状MoS2(如图2b所示);MoS2(002)晶面的间距为0.65 nm,如图2c所示。作为超级电容器的电极材料,花状MoS2球在3 A·g-1的大电流密度下,恒流循环充放电500圈后容量没有明显下降,比电容约为145 F·g-1。花状MoS2球作为超级电容器电极材料具有良好的循环性能和倍率性能的主要原因有:① 花状MoS2球的比表面积较高(8.7 m2·g-1);② 循环充放电过程中花状结构稳定存在。与2H-MoS2相比,1T-MoS2具有较大比表面积、良好的导电性以及亲水性。Xie等[22]通过水热法合成碳量子点(carbon dots,CDs)和1T-MoS2的复合材料,即CDs@1T-MoS2。研究表明,碳量子点能够诱导形成1T-MoS2,柠檬酸的用量对复合材料的形貌有重要影响。将该复合材料作为超级电容器的电极材料,在电流密度为0.5 A·g-1的测试条件下,其比电容为380 F·g-1,经过2000次充放电循环后容量保持率高达97%。
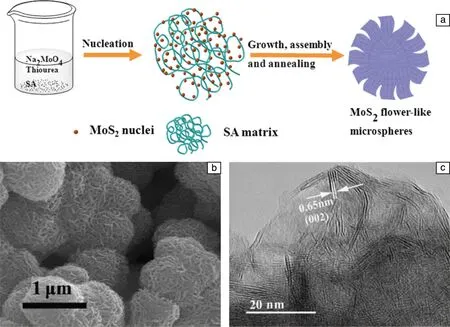
图2 海藻酸钠辅助水热法合成花状MoS2示意图(a);花状MoS2 SEM和TEM照片(b,c)[21]Fig.2 Illustration of the formation process of the flower-like MoS2 microspheres (a);SEM and TEM images of flower-like MoS2(b,c)[21]
为了进一步提高MoS2的电化学性能,往往将它与碳材料复合,以提高电极材料导电性,增加电极材料的稳定性[23]。Yang等[24]以SiO2球为模板,制备了空心碳球(hollow carbon spheres,HCS),通过水热法在空心碳球上制备了纳米MoS2,形成了具有层状介孔结构的MoS2@HCS复合材料。该复合材料比表面积高达119.0 m2·g-1,为离子的传输提供了丰富的通道;空心碳球能够提高复合材料的导电性,并且作为超级电容器的电极材料能够在充放电过程中保持稳定。Sangabathula等[25]采用了更简单的方法制备了纳米MoS2与碳的复合材料。在水热反应过程中加入蜡烛燃烧灰作为碳源,钼酸铵为钼源,硫脲为硫源,制备了MoS2/C复合材料。在20 A·g-1的电流密度下,循环10 000圈后,比容量保持率为94.8%。
在纳米MoS2材料加入碳材料也能够提高它作为电池电极材料的性能[26]。Ren等[27]在石墨烯泡沫(graphene foam,GF)和碳纳米管(carbon nanotube,CNT)组成的复合材料上制备了MoS2复合材料GF@CNT@MoS2。制备流程如图3a所示:先用化学气相沉积法在泡沫镍表面沉积了一层石墨烯;然后去除泡沫镍模板,在石墨烯表面涂覆了碳纳米管,形成GF@CNT;最后以硫代钼酸铵为原料用水热法在GF@CNT表面合成MoS2,得到GF@CNT@MoS2复合材料,其形貌如图3b~3e所示。虽然MoS2活性较高,但其导电性不及石墨烯和碳纳米管。实验结果表明:硫代钼酸铵的用量决定了复合材料中的MoS2含量,加入50 mg硫代钼酸铵制备的GF@CNT@MoS2-50既发挥了石墨烯和碳纳米管的导电性,也获得了较高的比容量和良好的循环稳定性。循环充放电测试结果如图3f所示,在0.2 A·g-1电流密度下循环25圈之后,复合材料GF@CNT@MoS2-50的比容量几乎不变,具有较高的可逆性。Wan等[28]以片状Mg(OH)2为模板,蔗糖为碳源,采用水热法合成了具有蠕虫状结构的C@MoS2纳米球。这种疏松的结构具有很高的比表面积,有效提高了C@MoS2纳米球作为锂电池负极活性物质的容量与循环稳定性。

图3 GF@CNT@MoS2制备原理示意图(a)和SEM形貌(b~e);0.2 A·g-1电流密度下GF@CNT@MoS2电极的循环性能(f)[27]Fig.3 Preparing process illustration (a)and SEM morphologies (b~e)of the GF@CNT@MoS2 hybrid foam;cycle performance of GF@CNT@MoS2 electrode at a current density of 0.2 A·g-1(f)[27]
近年来可穿戴设备逐渐成为各大厂商追逐的热点领域,而柔性电池是可穿戴设备必不可少的技术。Lin等[29]通过图4a所示的策略制备了柔性锂电池的电极材料SnS2/MoS2/CFC,制备方法大致可分为3步:首先,在活化后的碳纤维布(carbon fiber cloth,CFC)上通过水热法制备MoS2/CFC复合材料;然后在惰性气氛下煅烧,使亚稳相1T-MoS2转化为2H-MoS2;最后,通过溶液反应法,在MoS2/CFC复合材料表面生成SnS2,得到SnS2/MoS2/CFC复合材料。如图4g所示,SnS2纳米粒子增加了复合材料与电解液的接触面积,显著缩短了锂离子的扩散长度;MoS2带来较高的比容量;呈网状的碳纤维为电子的快速传输提供了路径。由于独特的结构设计,SnS2/MoS2/CFC展现出了稳定的循环性能(图4e)和低阻抗(图4f)的特性。在充放电电流密度为100 mA·g-1,循环充放电120圈后,可逆容量高达1294 mAh·g-1。
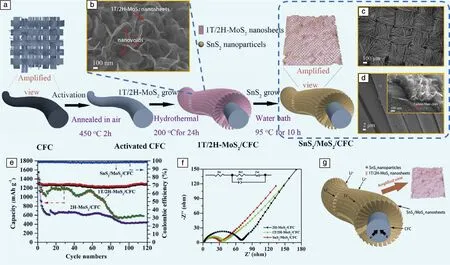
图4 SnS2/MoS2/CFC柔性电极制备流程示意图(a);1T/2H-MoS2(b)、CFC(c)和SnS2/MoS2/CFC(d)的SEM形貌;1T/2H-MoS2/CFC,2H-MoS2/CFC 和 SnS2/MoS2/CFC电极在100 mA·g-1下的循环充放电性能(e)和交流阻抗谱(f);SnS2/MoS2/CFC中的锂离子插入路径及电子传输路径的示意图(g)[29]Fig.4 Schematic diagram of the preparation of SnS2/MoS2/CFC flexible electrode (a);SEM morphologies of 1T/2H-MoS2 nanosheets (b),CFC (c)and SnS2/MoS2/CFC (d);cyclic charge-discharge performance (e)and AC impedance spectra (f)of 1T/2H-MoS2/CFC,2H-MoS2/CFC and SnS2/MoS2/CFC electrode at 100 mA·g-1;schematic diagram of lithium ion intercalation path and electron transport path in SnS2/MoS2/CFC (g)[29]
纳米MoS2不仅可以应用于常规的锂离子电池电极材料,还可以抑制穿梭效应,能够促进多硫化锂向最终的放电产物转化,提高锂硫电池的循环稳定性和可逆容量。Zhang等[30]以钼酸铵、硫脲、醋酸镍为原料,采用水热法制备了复合材料Ni-MoS2/rGO,并与S(硫单质)再复合制备了S-Ni-MoS2/rGO,测试了该材料作为锂硫电池正极材料的性能。结果表明,在充放电过程中,MoS2不仅能够吸附多硫化物,抑制“穿梭效应”,还能催化多硫化物向最终放电产物转化。Zhang等[31]通过第一性原理计算表明MoS2和MoS2-x(001)晶面能够促进多硫化物向硫化锂的转变。Lin等[9]通过实验证实了含硫缺陷的MoS2在锂硫电池中展现出优异的电化学性能。在倍率为0.5C的条件下,MoS2-x/rGO/S、MoS2/rGO/S的初始容量分别为1159.9 mAh·g-1和1033 mAh·g-1,循环150圈后的容量分别为819.9 mAh·g-1和576.4 mAh·g-1。Xiang等[32]先通过高温煅烧的方法制备了氮、硫共掺杂的碳纳米管(NSCNTs),然后采用水热法在NSCNTs表面合成了NSCNTs/MoS2复合材料。研究表明,NSCNTs/MoS2复合材料在充放电过程中,MoS2对多硫化物具有较强的吸附作用并且能够促进多硫化物向最终放电产物的转化,削弱了“穿梭效应”;NSCNTs为电子和锂离子的传输提供了快速通道,提高了锂硫电池的倍率性能。
在锌离子电池和钠离子电池中,纳米MoS2也有着广泛的应用。与锂电池相比,锌离子电池安全性高、成本低、对环境友好,具有广阔的应用前景。但锌离子的半径较大,不利于其在电极材料中嵌入和脱出。钠离子电池也存在类似的问题。钠离子的半径虽然小于锌离子的,但在充放电过程中也会使负极材料的体积发生变化,降低了电池的可逆比容量和循环稳定性。因此,扩大层状材料的层间距能够提高锌离子电池和钠离子电池的可逆比容量,降低容量衰减[33,34]。
氧掺杂不仅能增加活性位点的数量,还能增加MoS2的层间距,提高锌离子嵌入/脱出过程中的结构稳定性[35]。Li等[36]通过一步水热在碳布表面制备了含有氧原子掺杂的MoS2复合材料。研究表明,氧掺杂后纳米MoS2的层间距由0.62 nm增加到了0.96 nm。密度泛函理论计算表明,氧掺杂降低了锌离子迁移的能垒,提高锌离子的迁移速率。此外,氧掺杂还提高了纳米MoS2的亲水性。以该氧掺杂MoS2制备的水系锌离子电池的充放电测试结果表明,在0.1 A·g-1的电流密度下,比容量达到了208.5 mAh·g-1;在5 A·g-1的大电流密度下,比容量可达73.9 mAh·g-1。Huang等[37]也采用增加MoS2层间距的方法来提高水系锌离子电池的性能。在水热过程中加入葡萄糖作为插层剂,羧基化的碳纳米管作为导电骨架,制备了MoS2@CNTs复合材料。研究表明,在电流密度为1.0 A·g-1下,纳米MoS2的层间距从0.62 nm增加到了0.96 nm。充放电测试表明,比容量可达111.7 mAh·g-1,循环500圈后,容量保持率为80.1%。

此外,还可以通过构筑特殊的结构[41]来提高MoS2及其复合材料导电性和离子的传输速率。聚吡咯(PPy)、聚苯胺(PANI)、聚噻吩(PTh)等导电聚合物不仅可以构筑各种特殊结构,还可以提高电极材料的导电性。Wang等[42]以FeOOH@C为模板,通过水热法合成了C@MoS2@PPy复合材料,合成路径如图5所示。该复合材料具有中空结构,中空结构能够缓解钠离子嵌入、脱出而产生的体积变化,还能为离子的传输提供通道;导电聚合物PPy提高了材料的导电性。充放电测试表明,在0.1 A·g-1的电流密度下,循环100圈后,可逆比容量高达713 mAh·g-1;5 A·g-1的大电流密度下,循环500圈后,可逆比容量为294 mAh·g-1。与上述案例类似,Yu等[43]先通过水热法在碳布上制备了纳米MoS2,再利用吡咯的聚合反应形成了MoS2-PPy电极。3D结构的碳布缓解了电极的体积膨胀,碳布和PPy提供了导电网络,提高了电极循环稳定性。与未添加PPy电极相比,PPy缓解了电解液对纳米MoS2的腐蚀,电极循环稳定性明显提升。
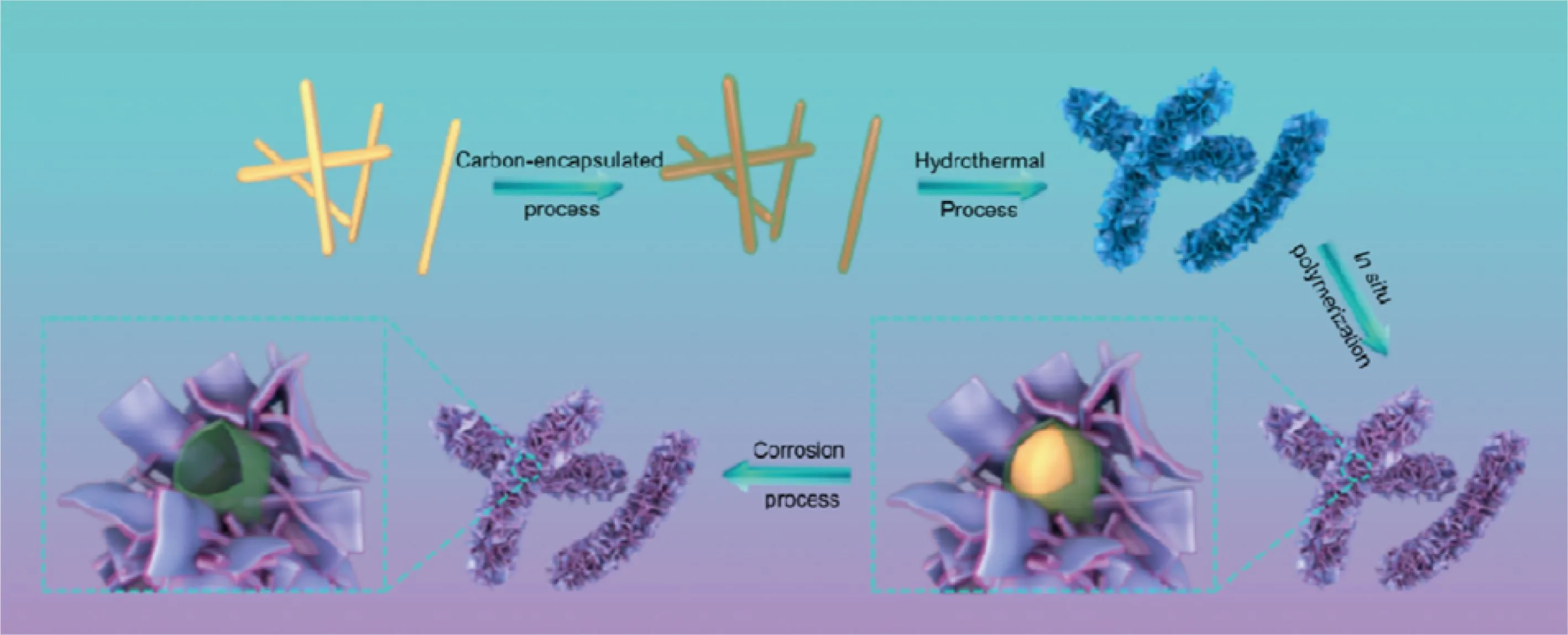
图5 C@MoS2@PPy的制备过程示意图[42]Fig.5 Schematic diagram of preparation process of C@MoS2@PPy[42]
总之,水热法合成纳米MoS2及其复合材料在储能领域有着广泛的应用。从提高导电性的角度出发,可采用与碳材料复合、诱导形成1T相MoS2、原子掺杂等方式。从提高循环稳定性的角度出发,可采用插层、原子掺杂等方法增加层间距,缓解充放电过程中的体积变化。
3.2 光催化领域
MoS2具有载流子迁移率高、对电子和空穴复合有较强的抑制作用、光敏性好、在可见光到近红外(NIR)区光吸收良好等独特性能。Thanga Gomathi等[44]通过两步水热法,以纤维素纸为载体制备了ZnS-MoS2复合材料,其制备工艺流程如图6a所示。首先,以硫脲、钼酸钠为原料,采用水热法在纤维素纸表面合成制备了一层花状MoS2。再将上述纤维素纸放入硫脲和氯化锌的溶液,通过水热法得到ZnS-MoS2-纤维素纸复合材料。如图6b~6e所示,ZnS呈实心球状均匀分布在MoS2之上,MoS2片状聚集成球形附着于纤维素纸的表面。ZnS与MoS2的能带结构存在差异,在复合材料界面处产生内建电场(如图6f所示)。在紫外光的作用下,ZnS中产生光生电子空穴对;在可见光和红外光作用下,MoS2中产生光生电子空穴对。由于复合材料在界面处存在内建电场,ZnS中的光生电子迁移到MoS2的导带,MoS2中的光生空穴迁移到ZnS的导带,避免和光生电子空穴对的复合,提高了光催化的效率。

图6 两步水热法制备ZnS-MoS2-纤维素纸复合材料的工艺流程图(a);MoS2-纤维素纸(b,c)和ZnS-MoS2-纤维素纸复合材料(d,e)的SEM形貌;光照下ZnS-MoS2界面能带及电荷转移机理示意图(f)[44]Fig.6 Schematic diagram of the two-step hydrothermal process for the fabrication of ZnS-MoS2-cellulose paper composite(a);SEM morphologies of MoS2-cellulose paper (b,c)and ZnS-MoS2-cellulose paper composite (d,e);energy band diagram of ZnS-MoS2 interface and schematic diagram of charge transfer mechanism occurred in ZnS-MoS2 under photoillumination(f)[44]
利用MoS2边缘对氧气的吸附特性,Li等[45]制备了复合材料Cu2O-MoS2/graphene(CMG),用于光催化降解有机物。制备方法如图7a所示,首先采用水热法在石墨烯表面合成了MoS2,然后在其表面沉积一层Cu2O,形成复合材料CMG,其微观形貌如图7c和7d所示。在复合材料Cu2O/graphene(CG)、Cu2O/MoS2(CM)和CMG中,复合材料CMG的光致发光强度最低(图7b),这表明复合材料CMG中光生电子空穴对的分离效率较高。MoS2和石墨烯促进了光生电荷的有效分离,延长了载流子寿命,阻碍了光生电子-空穴对的复合。复合材料CMG的光催化机制如图7e所示:Cu2O中的价带电子在可见光的作用下跃迁到导带,通过石墨烯迁移到MoS2表面,与吸附在MoS2边缘Mo上的氧气结合,形成超氧阴离子自由基。Cu2O中的空穴和MoS2边缘的超氧阴离子自由基吸收N-芳基-四氢异喹啉中氮原子的孤对电子,使含氮基团带正电,与亲核试剂发生反应。在整个反应中,N-芳基-四氢异喹啉被氧化,氧气被还原。

图7 CMG光催化剂合成示意图(a);Cu2O、CG、CM和CMG的光致发光谱(b);CMG的TEM照片(c,d);CMG的光催化原理示意图(e)[45]Fig.7 Illustration of the Cu2O-MoS2/graphene (CMG)photocatalyst synthesis (a);photoluminescence spectra of Cu2O,CG (Cu2O/graphene),CM (Cu2O/MoS2)and CMG (b);TEM images of CMG (c,d);photocatalysis mechanism schematic diagram of CMG (e)[45]
g-C3N4具有较强的光吸收能力,是一种很有潜力的光催化剂。但它的光催化性能由于电荷的快速复合受到抑制。构建异质结能够提高光生电子空穴对的分离效率[46]。Liu等[47]设计合成了复合材料MoS2nanostructured-C3N4(MSNS-CN),有效阻止了光生电子和空穴的结合。合成过程如图8a所示,首先采用水热法合成了花状纳米MoS2,采用尿素热解法制备了g-C3N4,然后采用超声波化学法合成了MoS2-C3N4复合材料。MoS2-C3N4的光催化机理如图8b所示,在光的照射下,C3N4的光生电子跃迁到导带,光生电子会向导带能量更低的MoS2迁移,从而有效阻止了光生电子和空穴的结合。MoS2纳米片边缘不饱和的S原子吸引溶液中带正电的H+,并将光生电子转移给H+形成H2。

图8 MoS2-C3N4复合材料合成过程(a),g-C3N4、0.5%MSNS-CN的能带结构图与g-C3N4/MoS2异质结光催化析氢机理示意图(b)[47];LaNiO3/g-C3N4/MoS2纳米复合光催化体系反应机理示意图(c)[50]Fig.8 Synthesis process of MoS2/g-C3N4 nanocomposites (MSNS-CN)(a),energy band structure diagram of g-C3N4 and 0.5% MSNS-CN,and illustration of proposed mechanism of photocatalytic H2 evolution over g-C3N4/MoS2 heterojunction (b)[47];schematic illustration of the proposed reactions mechanisms for the LaNiO3/g-C3N4/MoS2 nanocomposite photocatalytic system (c)[50]
形成异质结能够提高复合材料的光催化性能。Jia等[48]采用水热法合成了MoS2/ZnS/ZnO-twigs复合材料。在复合材料中,MoS2/ZnS呈花球状分布于树枝状的ZnO表面。研究表明,MoS2/ZnS形成的异质结有效地增强光生载流子的分离;MoS2和ZnS分别与ZnO形成了Ⅱ型异质结,促进了载流子的迁移。Shan等[49]通过水热法制备了MoS2-SnO2纳米复合材料。研究发现,在复合材料中,MoS2与SnO2有一定的位向关系,形成了大量的异质结,这些异质结起到了电子转移隧道的作用,大大提高了复合材料光催化活性。研究表明,所制备的SnO2纳米颗粒的尺寸对形成有效的异质外延结构起着至关重要的作用。SnO2纳米颗粒的尺寸越小,MoS2外延生长形成异质结的几率就越高。
Bao等[50]采用多步水热法制备了LaNiO3/g-C3N4/MoS2复合材料,通过精确调控界面相互作用,建立了有效的Z构型异质结结构(如图8c)。在光照下,LaNiO3和g-C3N4中出现光生电子空穴对。由于LaNiO3和g-C3N4构成了Z型异质结,LaNiO3中的导带光生电子会迁移到g-C3N4的价带。经过逐层转移,光电子积累在MoS2上,空穴积累在LaNiO3价带上。MoS2表面丰富的活性中心催化Cr(VI)和H2O还原为Cr(III)和H2。在模拟太阳光下,产氢速率高达45.1 μmol·h-1,是g-C3N4的19.1倍、LaNiO3/g-C3N4的4.9倍。
Wu等[51]制备了GO/g-C3N4/MoS2复合材料,利用它们之间的异质结来提高光催化效率。其合成方法如图9a所示,将配置好的混合溶液转移到反应釜中,一步水热法合成GO/g-C3N4/MoS2复合材料。通过电子显微镜观察到,MoS2均匀地分布在片状g-C3N4上,片状的g-C3N4均匀地分布在石墨烯上。如图9b所示,g-C3N4导带中的光生电子通过石墨烯迁移到MoS2导带,MoS2价带中的空穴迁移到g-C3N4价带,从而实现了光生电子空穴对的分离。MoS2导带上的光生电子还原Cr(VI),g-C3N4价带上的空穴氧化染料。
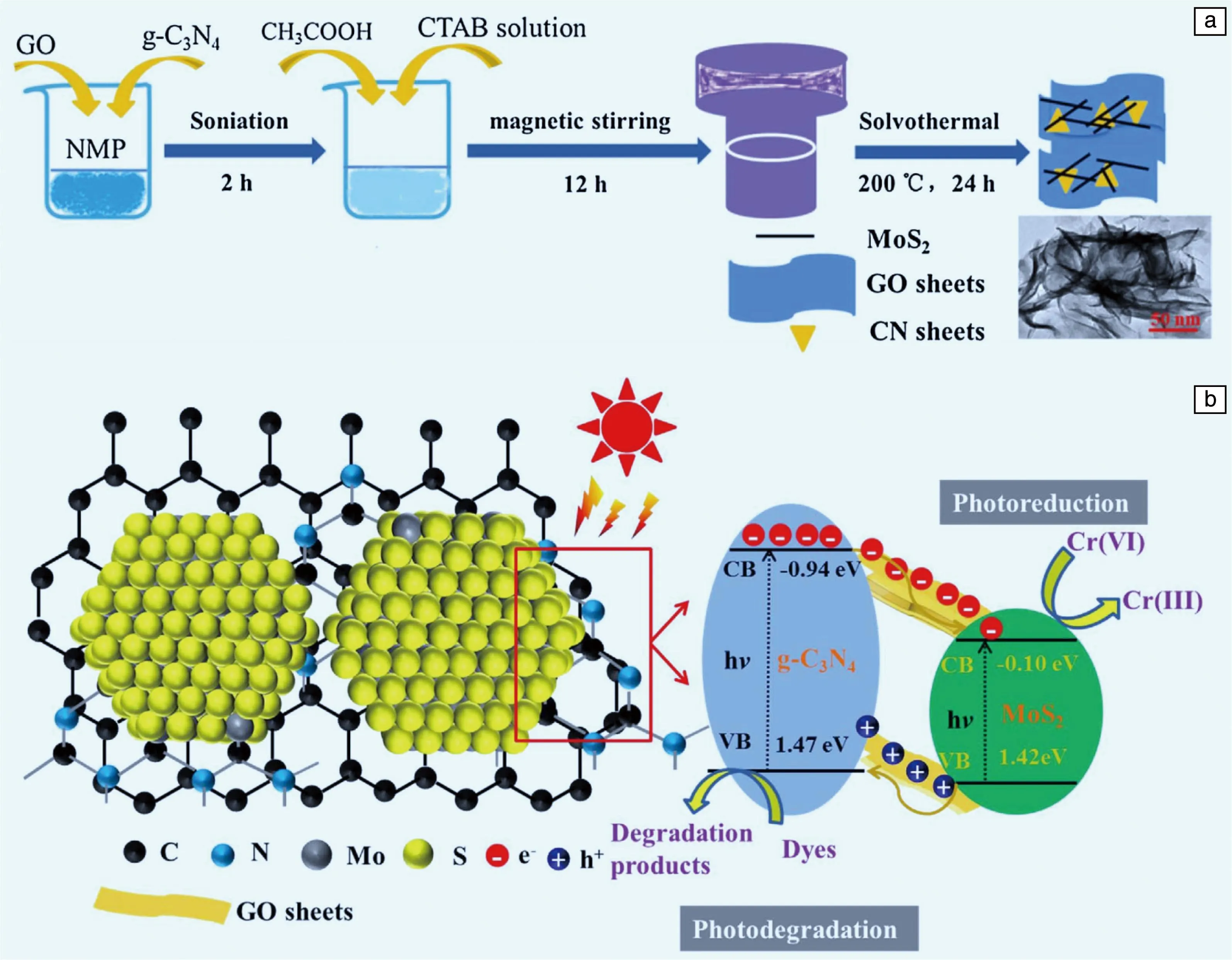
图9 GO/g-C3N4/MoS2复合材料合成过程示意图(a);GO/g-C3N4/MoS2复合材料光催化过程和电荷转移机理示意图(b)[51]Fig.9 Schematic diagram of the preparation procedure for GO/g-C3N4/MoS2 composite (a);schematic diagram of the photocatalytic process and charge transfer mechanisms of the GO/g-C3N4/MoS2(b)[51]
综上,提高纳米MoS2光催化性能的方法主要有:① 与具有光催化活性的物质构建异质结,抑制光生电子空穴对复合,提高光生电子空穴寿命;② 增加MoS2的活性位点,提高催化剂与被催化物质之间的离子迁移效率。
3.3 电催化析氢领域
金属Pt具有独特的电子结构以及高稳定性[52],是最佳的HER催化剂。然而Pt的储量少,价格高,阻碍了HER技术的发展。水热法制备的MoS2及其复合材料能有效提高电催化HER的效率,是替代金属Pt的材料之一。
通过优化MoS2的结构与形貌,提高层状结构边缘含有悬空键的Mo与S原子的比例,进而增加活性位点的含量,可提升电催化HER效率。Gao等[53]在水热反应过程中加入SiO2球作为硬模板合成了具有分级孔结构的MoS2球。在起始过电位为87.0 mV时,Pt/C电极的塔菲尔斜率为30 mV·dec-1;同样的条件下,MoS2球的塔菲尔斜率为48.8 mV·dec-1。由此可见,MoS2球具有良好的电催化析氢性能。van Tuan等[54]通过控制水热反应溶液的酸碱度,制备了MoS2纳米颗粒(MoS2-NP)、纳米花(MoS2-NF)、纳米球(MoS2-NS)、纳米中空结构(MoS2-NH)和MoS2/MoO2复合材料。电催化析氢测试结果表明,MoS2-NH的性能最佳,其过电位仅为230 mV(电流密度为10 mA·cm-2时),塔菲尔斜率为64 mV·dec-1。相关测试结果表明,空心的MoS2-NH的比表面积高达22.59 m2·g-1,在电催化过程中能够吸附H+并将其还原,缩短了离子传输的距离。
1T-MoS2具有良好导电性,在水热反应中诱导生成1T-MoS2能够提升离子迁移的速率。Zhang等[55]在水热合成MoS2的过程中加入溴化正丁基吡啶([BPy]Br)作为结构导向剂,制备了1T/2H-MoS2。[BPy]Br的空间位阻效应和π-堆积相互作用,诱导了MoS2从2H相向1T相的转变。1T/2H-MoS2在层间和边缘暴露了更多的活性位点,促进了电催化析氢过程中的电荷转移。电催化析氢测试结果表明,1T/2H-MoS2的塔菲尔斜率仅为59 mV·dec-1。Zhou等[56]通过提高水热反应中硫源的比例,诱导生成了层状的球形1T-MoS2。与2H-MoS2相比,1T-MoS2具有更加丰富的活性位点,1T-MoS2电极的电荷迁移率更高。电催化析氢测试表明,在同样的测试条件下,2H-MoS2电极的塔菲尔斜率为172.2 mV·dec-1,而1T-MoS2电极的塔菲尔斜率仅为96.4 mV·dec-1。
纳米MoS2中的缺陷也能够提升电催化析氢性能[57]。Dimakis等[58]通过理论计算发现,当存在S缺陷时,水分子及其解离组分H+和OH-才会强烈吸附在Mo原子周围。此外,在缺陷区域引入了较小的中间禁带,增加了MoS2的电导率,从而提高了电催化过程中电荷转移的效率。氧掺杂不仅能增加活性位点的数量,还能提高电催化活性[59,60]。Wu等[61]还通过控制水热合成中硫源与钼源的比例,合成了含有氧缺陷的二硫化钼(O-MoS2)。掺杂的氧和超过化学计量数的硫源赋予了MoS2大量的缺陷。这些缺陷提供了足够的活性中心,增强了对HER反应的催化能力。当硫脲与钼酸钠的物质的量比例为40∶1时,合成的O-MoS2电催化性能最佳。它与乙炔黑构成的电极电催化的起始电位为90 mV,Tafel斜率仅为47.24 mV·dec-1,在250 mV过电位下的电流密度达到了103.62 mA·cm-2。Liao等[62]的研究表明,Se原子的掺杂也能够提高MoS2电催化析氢的效率。他们通过两步水热法合成了Se-MoS2/Co0.2Ni0.8Se2复合材料,并研究了它的电催化性能。先采用水热法以泡沫镍为模板,制备了Co0.2Ni0.8Se2三维阵列,然后通过水热法在其表面制备Se-MoS2得到Se-MoS2/Co0.2Ni0.8Se2复合材料。测试表明,当电流密度为10和100 mA·cm-2时,过电位分别为30和122 mV。Co0.2Ni0.8Se2三维阵列具有良好的导电性和较大的比表面积,Se的加入使得纳米MoS2暴露出更多的活性位点。
为了使纳米MoS2的良好性能得到充分发挥,通常将纳米MoS2作为活性位点分布于碳材料之上,促进电催化析氢过程中的电子和离子的传输[63]。Zheng等[64]在水热合成中加入了SiO2球作为模板,合成了石墨烯与MoS2复合的中空纳米球,即h-rGO@MoS2。水热合成的纳米MoS2的比表面积为31.5 m2·g-1,而h-rGO@MoS2的比表面积高达144.9 m2·g-1,在电催化析氢过程中提供了更多的活性位点。在酸性介质、10 mA·cm-2的电流密度下,h-rGO@MoS2的过电位仅为76 mV,塔菲尔斜率为105 mV·dec-1。Yang等[65]采用水热法在碳布上合成了MoS2纳米片阵列,通过NH4F刻蚀获得含有氧的MoS2(O-MoS2)纳米片阵列。研究表明,O-MoS2中的不饱和硫原子为电催化的活性位点。增加刻蚀温度可增加该活性位点数量,提高电催化活性。与此同时,随着刻蚀温度的升高,O-MoS2的电荷转移电阻逐渐增加,不利于电催化。因此,需要合理选择刻蚀温度。电化学析氢性能测试表明,当刻蚀温度为120 ℃时,O-MoS2表现出良好的电催化性能,其塔菲尔斜率仅为49 mV·dec-1。Luo等[66]采用水热法在孔径150 μm的钛网上制备了一层花状MoS2,这种复合结构显著降低了电荷转移电阻,塔菲尔斜率只有30 mV·dec-1。掺氮的碳导电骨架(N-doped C matrix,NCM)具有良好的导电能力,不仅能够提高电极材料的导电性,还能提高循环过程中材料的结构稳定性。Shah等[67]以硫代钼酸铵((NH4)2MoS4)为前驱体,通过水热法制备了FeNi@NG/NCM@MoS2复合材料。在复合材料中,FeNi@NG附着在NCM表面,水热法合成的MoS2一部分位于NCM表面,另一部分位于FeNi@NG表面。测试表明,该复合材料具有良好的电催化析氢性能。在电流密度为10 mA·cm-2时,过电位为79 mV,塔菲尔斜率仅为40.2 mV·dec-1。第一性原理密度泛函理论模拟证实了电子从FeNi合金到FeNi@NG粒子表面的转移,并进一步转移到MoS2纳米片上,从而降低了Gibbs自由能,提高了HER活性。
综上,纳米MoS2制成的电极具有较低的塔菲尔斜率,纳米MoS2具有良好的电催化析氢活性。与储能领域的应用类似,提高导电性能够进一步提高电催化活性。此外,活性位点的含量也是决定电催化析氢性能的因素之一。
4 结 语
随着碳达峰、碳中和目标的提出,对储能材料、光催化以及电催化材料的需求越来越旺盛。水热法合成的纳米MoS2具有良好的电化学性能以及电催化和光催化性能。尽管纳米MoS2相关研究论文的数量逐年增加,但是水热法合成的纳米MoS2研究还存在以下两方面的问题:
(1)水热法合成纳米MoS2反应的历程尚不明确,可重复性差。反应历程的研究可为反应条件的设置以及前驱体的选择提供指导性意见。目前,水热反应难以直接观测,只能通过温度、压力的监测,结合反应物的特性,来推断反应的过程。在同样的条件下,产物的形貌有着较大的差异。
(2)水热法成本高,原料利用率低。水热法制备纳米MoS2的过程中,溶液浓度低,废水中含有未形成沉淀的金属离子,存在污染环境的风险。
在水热法合成MoS2后续的研究中,既要注重反应历程的研究,还要积极探索反应废水循环工艺。在反应历程研究时,应模拟反应釜中的传质传热过程,结合前驱体的反应特性以及温度压力,更精准地描述反应的历程。在工艺方面,根据水热反应废水的成分、pH、产量,适当调节后,将废水作为反应的溶剂重复利用。这些研究将降低水热法合成纳米MoS2的成本,提高不同批次产物的一致性。随着技术的发展,不同形貌和特性的纳米MoS2将应用于人们生活的各领域中。
