基于多层注意力和消息传递网络的药物相互作用预测方法
2024-01-09饶晓洁孟献兵陈俊龙
饶晓洁 张 通 ,2 孟献兵 陈俊龙 , 2
药物相互作用(Drug-drug interaction,DDI)针对的是两种或两种以上药物进行混合时,某些药物的性能受到其他药物的影响,而发生协同或拮抗作用[1-2].DDI 引发的药物副作用可能会降低药物疗效,诱发不良反应,甚至影响到患者的身体健康.由于传统基于实验的方法进行DDI 预测存在成本高、检测周期长等问题[3-4],近年来,基于计算机辅助的计算方法日益得到广泛应用[2,5].通过传统的机器学习和深度学习等计算机辅助计算方法从已有药物相互作用数据中学习建立模型并实现DDI 预测,可以大大提高DDI 预测任务的效率[3].因此,研究基于计算机辅助的DDI 预测方法具有重要的理论和应用价值.
DDI 预测任务主要包括对药物分子式进行量化并提取其特征,以及选择合适的模型预测DDI[6-7].不同于一般的结构化数据,药物分子具有确定的分子结构及其生物化学性质.因此,解决DDI 预测问题的关键是学习药物分子结构及其特征信息,并建立预测模型.目前,已有大量DDI 预测任务相关研究成果,其中涉及的方法可以概括为基于传统机器学习的方法和基于深度学习的方法[1,8-9].
在现有DDI 预测方法中,利用药物的生化特征信息,包括药物靶点、酶、药物转运体及蛋白质等相关信息构建模型并预测DDI,是一种有效的研究思路[2,10-11].这类方法通过充分利用药物相关联的生化特征信息可以提高DDI 预测的精度,不过也存在一些局限性[3],比如此类生化特征信息的获取成本较高[6].如何在没有这些生化特征信息的前提下提高DDI 预测的精度,是值得深入研究的问题.此外,由于DDI 预测任务涉及多个药物分子,且不同药物分子内部又具有不同的原子信息,因此有必要深入挖掘药物分子内不同原子和不同药物分子之间的相关关系,并研究通过融合原子和分子等不同层次的特征信息,提高DDI 预测准确率.
针对上述问题,本文提出一种基于多层次注意力机制和消息传递神经网络的药物相互作用预测方法.为了充分挖掘药物的分子结构信息,本文从药物分子内不同原子和不同药物分子之间两个层面研究DDI 预测方法.通过基于注意力机制的消息传递神经网络学习药物分子内包含不同原子和化学键的图结构特征,并结合基于多头注意力机制的神经网络提取不同药物分子间相关关系的特征信息,实现从药物分子内和分子间两个不同层次进行药物分子特征提取,从而完成DDI 预测任务.
本文主要贡献是同时从原子和分子层面设计基于注意力机制的特征学习策略,提出基于多层次注意力机制和消息传递神经网络的DDI 预测方法.主要创新点包括两个方面: 一方面,通过考虑不同原子及其相关联化学键的不同相互作用信息,设计基于分子质心的位置编码策略,提出基于注意力机制和消息传递神经网络的原子特征网络;另一方面,通过考虑分子之间的不同相互作用关系,提出基于注意力机制和密集连接结构的分子特征网络.具体来说,相比直接在分子层面学习不同药物分子相互作用关系的方法[10,12],本文方法选择同时从原子和分子层面学习预测DDI.考虑到不同药物分子的相互作用关系本质上取决于其内部原子和化学键的相互作用,这里首先学习药物分子内不同原子、化学键间的相互作用关系,并通过设计基于分子质心的位置编码策略,辅助学习药物分子的图结构特征表示.这种方法有助于模型学习得到丰富的药物分子表示,从而更有利于挖掘出不同药物分子的潜在相互作用关系.虽然已有研究工作也同时从原子和分子层面研究DDI 预测方法,不过这些方法在原子层面只考虑了原子节点特征,并没有充分利用不同原子相关联的化学键特征[13],无法在原子和分子层面同时提取不同原子及其相关联化学键的不同注意力信息,因此其预测能力也有限.大量对比实验和消融实验验证了本文方法的有效性,以及相比现有方法的优越性.
1 相关工作
基于计算机辅助计算的DDI 预测方法可以概括为基于传统机器学习的方法和基于深度学习的方法.基于传统机器学习的方法主要分为3 类,即基于传统分类器的方法和基于回归的方法和基于矩阵分解的方法[8,14-15].在基于传统分类器的方法和基于回归的方法中,通常会使用相似性度量方法(基于内积或基于余弦值的相似度度量)度量两种药物之间的相似度,并通过不同的分类或回归算法,预测得出不同药物相互作用的概率[8,14].在基于矩阵分解的方法中,DDI 预测任务可以建模为矩阵补全任务: 将一个含有缺失值的矩阵恢复为一个完全的矩阵,目的是对未观察到的相互作用进行预测[15].基于传统机器学习的方法虽然能有效解决DDI 预测任务,但因其提取深层特征的能力有限,也存在一定的局限性,比如在不平衡数据以及大规模数据集上存在表现能力不足的问题[5].此外,这类方法往往直接利用药物分子的特征信息,而忽略药物分子内部的原子特征信息,这在一定程度上也会影响DDI预测的效果.
不同于基于传统机器学习的方法,基于深度学习的DDI 预测方法能够提取更深层次的特征,在实际应用中,往往可以更好地预测出潜在的DDI.如,Ryu 等[1]提出了一种基于深度学习的DDI 预测模型,通过学习不同药物的结构相似度信息,实现DDI 预测.Deng 等[10]通过计算药物结构、基因本体和目标基因这三种相似度信息,结合深度神经网络,实现药物分子特征提取,并用于DDI 预测.Lee 等[6]利用每种药物不同的相似度信息训练模型,使用自动编码器和深度前馈网络实现DDI 预测.这些方法借助深度学习深层特征提取能力[1,6,10,12],虽然也能解决DDI 预测问题,但忽略了药物基于图结构的数据本质,实际应用中效果往往有限.
近年来,基于图结构的深度学习方法相继提出并成功应用于DDI 预测[5,9].这类方法通过将不同药物分子作为节点、相互作用关系作为边,构建药物分子的图网络,从而实现DDI 预测[7,16].如,Liu等[9]使用多模态深度自编码器,将每个药物数据源视为一个药物特征网络,在每个网络中利用图结构的邻接矩阵做图嵌入,从多个药物特征网络中学习药物的统一表示,并在此基础上构建模型,实现DDI 预测.Lin 等[16]设计出一种基于知识图谱的图卷积神经网络,通过学习基于不同药物分子的知识图谱,获取药物潜在的相互作用关系.Karim 等[7]提出一种DDI 预测模型,通过知识图谱学习药物的重要特征,并通过集成卷积神经网络和长短期记忆递归神经网络进行学习,得到不同药物的相互作用关系.这类方法虽然考虑了药物分子的图结构信息,但也存在一定的局限性.例如,上述方法从药物分子层面进行特征提取,而忽略了药物分子内原子层面的特征学习.此外,这些方法并没有考虑通过区分不同药物分子或药物原子的重要性来预测DDI.针对上述问题,本文提出了基于药物分子内和药物分子间的多层次注意力机制和特征提取方法.
2 本文方法
本文研究的DDI 预测任务是预测给定药物分子集合中任意2 个药物分子的相互作用关系.这里采用简化分子线性输入规范(Simplified molecular input line entry specification,SMILES)表示每个药物分子,并将DDI 任务建模为通过提取SMILES文本序列表示的药物分子特征预测得出两种药物是否存在相互作用的链接预测问题.理论上来说,不同药物是否存在相互作用关系取决于药物的分子图结构信息及其相关生化性质.因此,对于基于SMILES文本序列的DDI 预测方法来说,从序列中准确提取药物分子图结构信息对提高DDI 预测方法的精度具有十分重要的作用.为此,本文基于Transformer的注意力机制[17]和消息传递神经网络[18],设计基于多层次注意力机制和消息传递神经网络的DDI 预测方法,旨在实现分子图结构特征提取及DDI 预测.
本文方法整体框架如图1 所示.首先,从SMILES文本序列中提取药物分子的图结构信息,包括原子和化学键相关联的特征[19];然后,在原子特征层面,利用消息传递神经网络,并通过融合Transformer的注意力机制以及本文提出的基于分子质心的位置编码方法,实现药物分子内不同原子和化学键特征的学习更新;最后,在分子特征层面,进一步利用注意力机制,并通过监督学习和对比学习,挖掘出不同药物分子的潜在相互作用关系,从而实现任意两个药物分子i和j的相互作用预测.
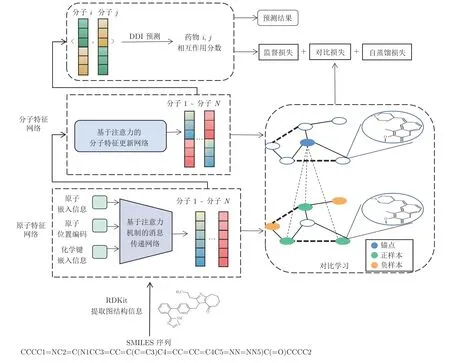
图1 模型框架图Fig.1 Framework of the proposed model
2.1 基于注意力机制的消息传递原子特征网络
药物分子内部由不同原子及原子之间相关联的化学键组成.对于具有图结构的药物分子来说,SMILES 这种一维线性的序列表示无法直接反映不同原子和化学键在药物分子图结构中的相对位置信息.为解决这个问题,并提取药物分子内不同原子和化学键的特征信息,本文建立基于注意力机制的消息传递原子特征网络,学习得到基于图结构的药物分子特征表示.
原子特征网络如图2 所示.首先,使用药物原子和化学键信息进行节点和边的特征嵌入,同时设计基于分子质心的位置编码方法编码具有图结构的原子和化学键特征信息;然后,利用结合图结构交互式注意力机制的消息传递神经网络,学习不同节点和边的特征信息;最后,通过不断更新迭代模型,得到药物分子的特征表示.下面具体从两个方面进行介绍.
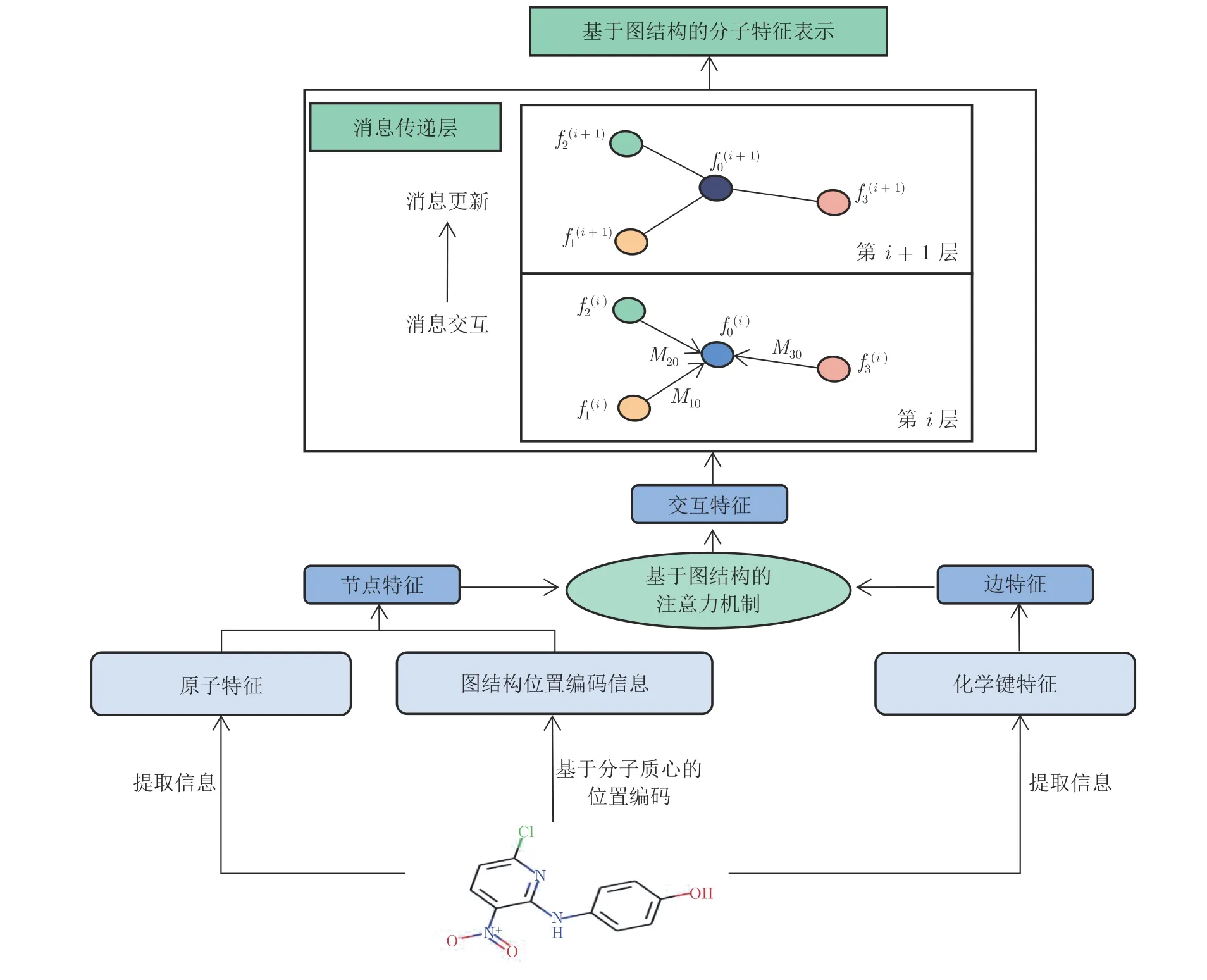
图2 基于注意力机制的消息传递原子特征网络Fig.2 Framework of the message passing atomic feature network base on attention mechanism
2.1.1 基于分子质心的位置编码
由于药物分子是不同原子和化学键组成的图结构数据,因此在Transformer 注意力机制中处理时序数据的位置编码方法不一定适合药物分子数据.此外,由于表示药物分子的SMILES 是一维线性化的序列,因此仅仅利用药物分子SMILES 序列中字符的输入先后顺序作为位置编码的依据[18]是不合理的.为了有效提取药物分子中不同原子的位置信息,本文提出一种基于分子质心的位置编码方法,通过计算原子与分子质心的距离来表示原子之间的相对位置,得到基于该距离的排序结果并用于设计位置编码.
给定具有n个原子的药物分子,该分子可表示为有向图G=(X,E),其中X,E是通过RDKit[19]化学信息库分别得到的原子和化学键的初始特征.X由药物分子内n个原子组成,表示原子节点i的特征嵌入信息,包括通过RDKit 获取得到原子的杂化方式、形式电荷、连接数等信息.E代表原子之间相关联的化学键,包括化学键的类型、是否为芳香键、是否成环等特征信息.euv ∈E ∈表示从原子节点i到节点j的边特征嵌入信息.fn,fe分别表示节点和边的特征维度.拓扑连接矩阵ADJ ∈Rn×n由两个原子之间的最短路径组成.基于分子质心的位置编码方法,具体如下:
首先,通过计算分子中n个原子二维坐标vi(i=1,2,3,···,n) 的均值v,得到分子质心的坐标s0;其次,计算每个原子和分子质心之间的欧氏距离di,再按照n个原子与分子质心s0之间的距离值di由近到远进行排序,得到距离分子质心由近到远的原子索引;最后,将该原子索引顺序作为n个原子的位置编码,经过词嵌入得到n个原子的位置编码posi.基于分子质心位置编码的原子特征h(xi) 和边特征h(euv) 表示方法分别为
这种位置编码方法通过利用分子图结构的空间信息描述不同原子的相对位置关系,在一定程度上可以改善分子SMILES 序列表示方法仅以SMILES 序列中字符输入先后顺序作为位置编码的不足,为原子的特征表示学习提供更多的信息.
2.1.2 基于注意力机制的消息传递神经网络
针对传统消息传递神经网络不能有效区分不同节点和边的不同作用信息的问题[13],本文借鉴基于图交互式的消息传递神经网络的思想[18],将药物分子中不同原子及其相关联的化学键(边)表示为区分入边和出边的有向图,利用基于Transformer 注意力机制的消息传递神经网络,结合本文提出的基于分子质心的位置编码策略,计算药物分子中不同原子和化学键之间相互作用的注意力分数,并进行基于图结构的消息传递,不断学习更新节点和边的特征,从而得到药物分子的特征表示.通过将药物分子表示为有向无环图,节点和边的信息只会沿着确定的方向传递,不会出现因无向图中的环路造成节点和边信息的循环更新问题,从而提高节点和边信息的学习更新效率.此外,由式(1)和式(2)可知,通过区分入边和出边,不同节点及其相关联的边将具有不同的信息.此时,通过基于注意力机制的图结构信息传递和更新学习,更容易学习得到不同原子及其相关联边的不同作用信息,从而提取到更有效的药物分子特征表示.
在基本消息传递神经网络中,通常利用邻接矩阵进行消息的聚合及更新[20].该方式存在只考虑节点特征而忽略边信息的缺点.同时,在进行消息传递时无法自适应调节节点之间信息传递权重.为了解决上述问题,本文引入了注意力机制.其中,计算Q,K,V的方式为
为了进一步挖掘图结构信息,本文将每个分子视为一个有向图,并将边分为入边和出边两个类型,利用式(3)求得的Q,K,V矩阵计算相应的消息传递分数矩阵Mi和Mo,即
其中,Mi,Mo ∈Rn×n,einsum 为爱因斯坦求和约定,Qi,Ki,Qo,Ko分别表示入边、出边相应的矩阵.最终的消息传递分数矩阵M为
其中,M∈Rn×n,s oftmax 将数值向量归一化为概率分布,d iag{Mo}表示只保留矩阵Mo的对角线元素.
考虑到在消息传递过程中,不同传递深度的消息携带的信息不同,为了模拟随着消息传递层数加深而导致的信息量减少的现象,本文引入消息衰减机制[18],并假设距离越远的两个原子之间交互分数衰减得越快.带有衰减机制的消息传递矩阵M计算式为
其中,γ为表示衰减程度的参数,ADJ(u,v) 表示原子u和v之间的最短路径.
经过单层消息传递后,得到的节点和边特征的更新式为
其中,matmul 表示矩阵乘法,⊙表示两个矩阵对应元素相乘.最后,本文使用平均池化的方式生成原子特征网络输出的分子表示,即
其中,Fa是所有分子特征组成的矩阵,Fai是第i个分子的特征表示,Ki表示第i个分子的最大原子数量,h(Xm)k表示经过上述网络更新后的第m个分子中第k个原子的特征.
2.2 基于多头注意力机制的分子特征网络
原子特征网络只考虑了单个分子内部的结构信息.如果直接基于原子特征网络输出的Fa进行DDI预测,将会丢失不同药物分子之间的交互信息,从而影响最终的DDI 预测结果精度.为了解决这个问题,本文将原子特征网络输出的分子表示作为分子特征网络的输入,通过设计多头注意力模块学习不同药物分子间的交互信息,并利用学习得到的药物分子关系更新每个药物分子的向量表示,最终基于不同药物分子的特征学习得到DDI 预测的结果.
通过对原子特征网络输出的药物分子特征表示Fa进行线性变换,得到Q′,K′,V′矩阵,并计算多头注意力,即
为防止网络层数过深导致的梯度消失,且实现不同层次特征的融合,本文在多层感知机(Multi layer perceptron,MLP)网络中线性层之间使用密集连接结构,计算最终的输出: 药物分子特征表示Fm.具体为
其中,x0是多头注意力层的输出,xl表示每个密集连接层的计算式,l ayerl表示第l个线性层,DenseMLPN表示一个具有N层密集连接的MLP 网络.
其中,E=Fm.
给定药物分子对i和j,通过式(13)计算得到Sij,再将其经过一个MLP 和sigmoid 函数,得到最终的链接预测结果pij.
本文模型的复杂度主要包括原子特征网络和分子特征网络以及对比学习这3 部分的计算.在原子特征和分子特征网络中主要计算量是式(8) 和(12),其相应的计算复杂度都是 O (NBf+Hd),其中,N指数据集中的药物分子数量,B是药物分子中的化学键数量,H表示能发生相互作用的药物对数量,f和d分别表示输入的特征维数和药物嵌入的特征维数.对比学习部分的计算复杂度是 O (Nk),其中k是正样本数量.因此,模型复杂度为O(N(Bf+k)+Hd).
2.3 模型的训练优化
为了提高模型的泛化性能,本文在传统二元交叉熵损失函数(Binary cross entropy,BCE)Llabel的基础上,引入2 种无监督损失函数,包括基于自蒸馏的正则化约束Lun和基于对比学习的无监督损失Lc.模型整体的损失函数L为
其中,α和β是对应损失的权重系数.
Llabel使用二元交叉熵损失函数衡量模型的误差损失,即
其中,Tr表示训练集的样本对集合,yij表示样本对 (i,j) 的真实标签,rij和pij分别表示原子特征网络和分子特征网络输出的预测结果.
对于基于自蒸馏的正则化约束[21],利用分子特征网络的输出对原子特征网络输出进行蒸馏学习,进一步提高原子特征网络输出特征的质量.这里通过KL 散度(Kullback-Leibler divergence,KL)表示Lun,即
其中,K L(pij ‖rij) 表示两个概率分布之间的 K L 散度,用来衡量两个分布之间的分布差异.DTr表示除训练集之外的样本对集合.
基于对比学习的无监督损失函数设计思想如下.对于每个药物分子,选取其在分子特征网络的输出特征作为锚点,并将其一阶邻居和非一阶邻居在原子特征网络的输出特征分别作为正样本和负样本.通过对比损失学习,使得锚点与其正样本相接近,与其负样本区分开来.具体为
其中,C(i) 和(i) 分别表示节点i的一阶邻居集合和非一阶邻居集合.ϕ,φ分别定义了原子特征网络和分子特征网络的参数,定义了互信息估计器.
由于无法直接优化互信息,本文使用JS 散度(Jensen-shannon divergence,JSD)优化互信息的下界[22].通过最小化对比损失函数Lc,使得互信息最大化,对比损失函数的计算式为
模型训练过程的伪代码如算法1 所示.
算法1.端到端的药物相互作用预测模型
3 实验结果及分析
为了验证本文方法的有效性和优越性,我们选择两个常用的DDI 数据集,即ZhangDDI[11]和ChCh-Miner[23],进行对比和消融实验分析.Zhang-DDI 包含548 种药物和48 548 组药物相互作用关系数据,ChCh-Miner 包含1 514 种药物和48 514组药物相互作用关系数据.评价指标包括ROC(Receiver operating charaeteristic curve)下面积(Area under ROC,AUROC)、PRC (Precision-recall curve)下面积(Area under PRC,AUPRC)和F1 分数(F1-score,F1).
3.1 对比方法介绍及实验设置
本文选择13 种具有代表性的DDI 预测方法作为对比方法,分为基于传统机器学习的DDI 预测方法和基于图结构的深度学习DDI 预测方法.
基于传统机器学习的DDI 预测方法包括以下6 种方法: 基于子结构相似性的DDI 预测方法NN(Nearest neighbor)[24];基于标签传播的DDI 预测方法,这里包括3 个基于不同相似性的方法(LPSub (Label propagation substructure)、LP-SE(Label propagation side effect)、LP-OSE (Label propagation off-label side effect))[25];基于混合集成模型的DDI 预测方法MF-Ens (Multi-feature ensemble)[11];基于结构相似性轮廓的DDI 预测方法SSP-MLP (Structural similarity profile and multilayer perceptron)[1].
基于图结构的深度学习DDI 预测方法又分为两类,即基于分子特征网络的DDI 预测方法、基于原子特征和分子特征网络的DDI 预测方法.第一类方法包括以下4 种方法: 基于图卷积网络的DDI 预测方法GCN (Graph convolutional network)[26]、基于图同构网络的DDI 预测方法GIN (Graph isomorphism network)[27]、基于图自动编码器的DDI预测方法Att-auto (Attentive graph autoencoder)[12]、基于图注意力网络的DDI 预测方法GAT (Graph attention network)[28].第二类方法包括以下3 种方法: 基于层次图表示学习的DDI 预测方法SEALCI (Semi-supervised hierarchical graph classification)[29]、基于分子指纹和图卷积网络的DDI 预测方法NFP-GCN (Molecular fingerprint graph convolutional network)[30]、基于键感知消息传递神经网络和图卷积网络的DDI 预测方法MIRACLE (Multiview graph contrastive representation learning)[13].
对于ZhangDDI 和ChCh-Miner 数据集,本文参照文献[13]的数据划分方式,所有数据样本按照4:1 的比例分为训练集和测试集,并在训练集中随机选择1/4 的样本作为验证集.当连续训练10 轮且模型在验证集上的最佳精度没有改变时,模型停止训练.所有实验结果都是通过5 次独立实验进行统计分析得到.在原子特征网络中,原子特征维度设置为115,化学键特征维度设置为13.在分子特征网络中,注意力头数设置为3.目标函数中的系数α和β分别设置为1 和0.8,实验基于Pytorch 1.6.0.
3.2 对比实验分析
在ZhangDDI 和ChCh-Miner 两个数据集上的实验结果分别如表1 和表2 所示.由表1 可知,相比于12 种对比算法,本文在所有指标上都取得最好结果.与基于键感知消息传递神经网络和图卷积网络的DDI 预测方法[13]相比,本文方法虽然在AUPRC 指标上取得次优结果,但是在AUROC 和F1 指标上表现更好、更鲁棒.由表2 的实验结果可知,当药物种类数显著增加时,本文方法超过了所有对比方法,且优势更加明显.
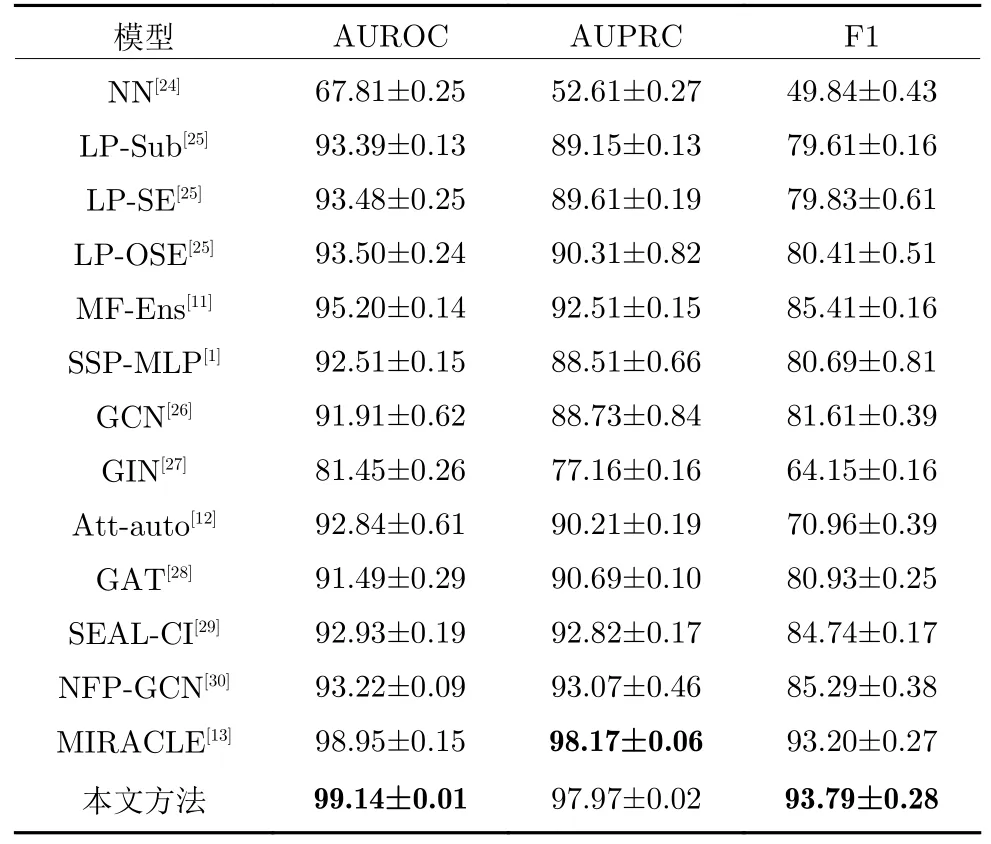
表1 ZhangDDI 数据集上的对比实验结果Table 1 Comparison experimental results on ZhangDDI dataset
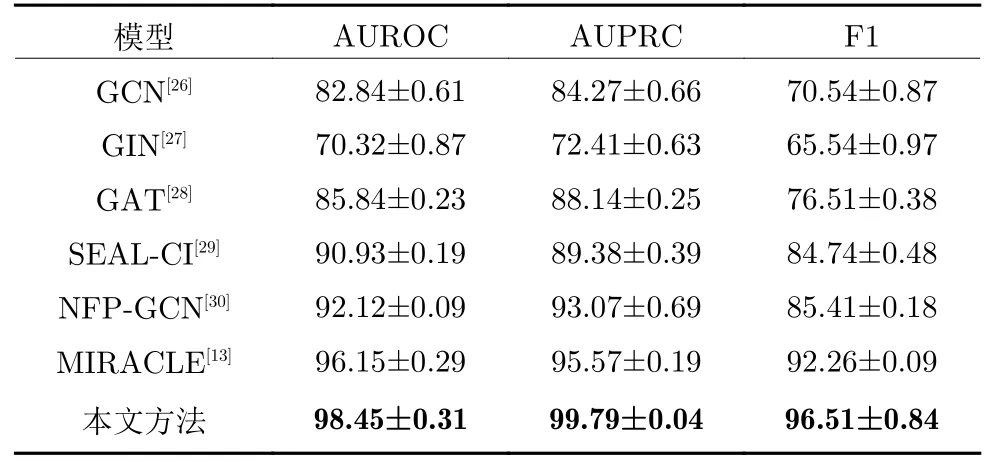
表2 ChCh-Miner 数据集上的对比实验结果Table 2 Comparison experimental results on ChCh-Miner dataset
1 )与6 种基于传统机器学习DDI 预测方法相比,本文方法在ZhangDDI 数据集上取得的3 项指标结果至少提高4%,5%,8%.这是由于基于相似性的DDI 预测方法是通过传统机器学习计算多种药物特征的相似度从而预测出DDI 结果,而药物分子往往具有复杂的结构特性,不能简单地由一种或几种特征刻画,且传统机器学习提取深层特征的能力有限,因此这类方法效果并不好.不同于此类方法,本文方法没有选择具体的药物分子特征,而是通过深度学习方法同时从药物原子和分子层面学习药物分子的深层特征.
2 )与4 种基于分子特征网络的DDI 预测方法相比,本文方法在ZhangDDI 数据集上取得的3 项指标结果至少提高6%,7%,12%,而在ChCh-Miner 数据集上,相应结果至少提高13%,12%,20%.基于分子特征网络的DDI 预测方法直接从药物分子层面学习不同药物分子的相互作用关系,忽略了分子内部的结构特性,算法性能受制于药物分子特征表示的好坏.而本文方法首先从原子层面学习得到每个药物分子的特征,然后结合监督学习和对比学习,不断优化得到的药物分子特征,并基于这些分子特征学习得到不同药物分子的相互作用关系,在理论上更具优势.实验结果也证明了本文方法的优越性.
3 )与3 种基于原子特征和分子特征网络的DDI预测方法相比,本文方法除1 个指标取得次优结果外,都能取得最好的结果.虽然这些方法都能从原子和分子层面提取药物分子特征,但它们都缺乏明确的机制学习原子和化学键之间、分子之间的不同注意力信息.例如,基于键感知消息传递神经网络和图卷积网络的DDI 预测方法 MIRACLE[13],虽然在AUPRC 指标上具有良好的竞争力,但其整体性能不如本文方法.这是因为MIRACLE 只考虑不同原子间的消息传递,并没有考虑边的特征信息.而本文方法可同时考虑不同原子及其相关边之间的消息传递,且可学习不同原子间的注意力信息.本文方法可以在原子和分子层面同时进行具有不同作用的注意力学习,因此,本文方法在综合性能表现上更优越.
3.3 消融实验分析
3.3.1 多层注意力机制的消融实验
为验证本文提出的多层次注意力网络的有效性,我们在两个数据集上针对基于注意力机制的原子特征网络和分子特征网络分别进行消融实验.
关于原子特征网络和分子特征网络的消融实验结果分别见表3 和表4.实验结果表明,无论是原子特征网络,还是分子特征网络,删除其注意力机制后,模型性能都会显著下降.如果缺乏基于注意力机制的原子特征网络,那么在最终的分子表示中将会丢失分子内部原子和边的特征信息,而这会直接影响药物分子的特征质量;同样,如果没有基于注意力机制的分子特征网络,那么将会丢失分子间的相互作用信息,导致模型只会根据两个独立的分子特征来进行DDI 预测.根据上述分析,本文提出的基于多层次注意力机制的原子特征和分子特征网络确实有助于提高药物分子的特征质量和模型性能.
为进一步验证本文提出注意力机制的有效性,我们将分子特征网络经过注意力机制计算得到的分子之间相互作用的注意力分数进行可视化.作为示例说明,这里展示在ZhangDDI 数据集上抽取的一个分子(记为A)和另外542 个分子之间的注意力分数,其中与A 发生相互作用和不发生相互作用的分子各占一半数量.图3 是分子A 与其他542 个分子之间的注意力分数经过归一化后的可视化结果,其中,图3(a)是与A 发生相互作用的药物分子的注意力分数可视化,图3(b)是不与A 发生相互作用的药物分子的注意力分数可视化.经过计算可得,与A 发生相互作用的药物分子的注意力分数之和占注意力分数总和的56.87%,平均注意力分数是0.21;而不与A 发生相互作用的药物分子的注意力分数之和则占注意力分数总和的43.13%,平均注意力分数是0.16.即当药物分子之间存在相互作用时,其注意力分数大于没有相互作用时的注意力分数.这在一定程度上说明通过本文提出的多层次注意力机制计算得到两个药物分子之间的注意力分数越大,则两个药物分子发生相互作用的可能性也越大.
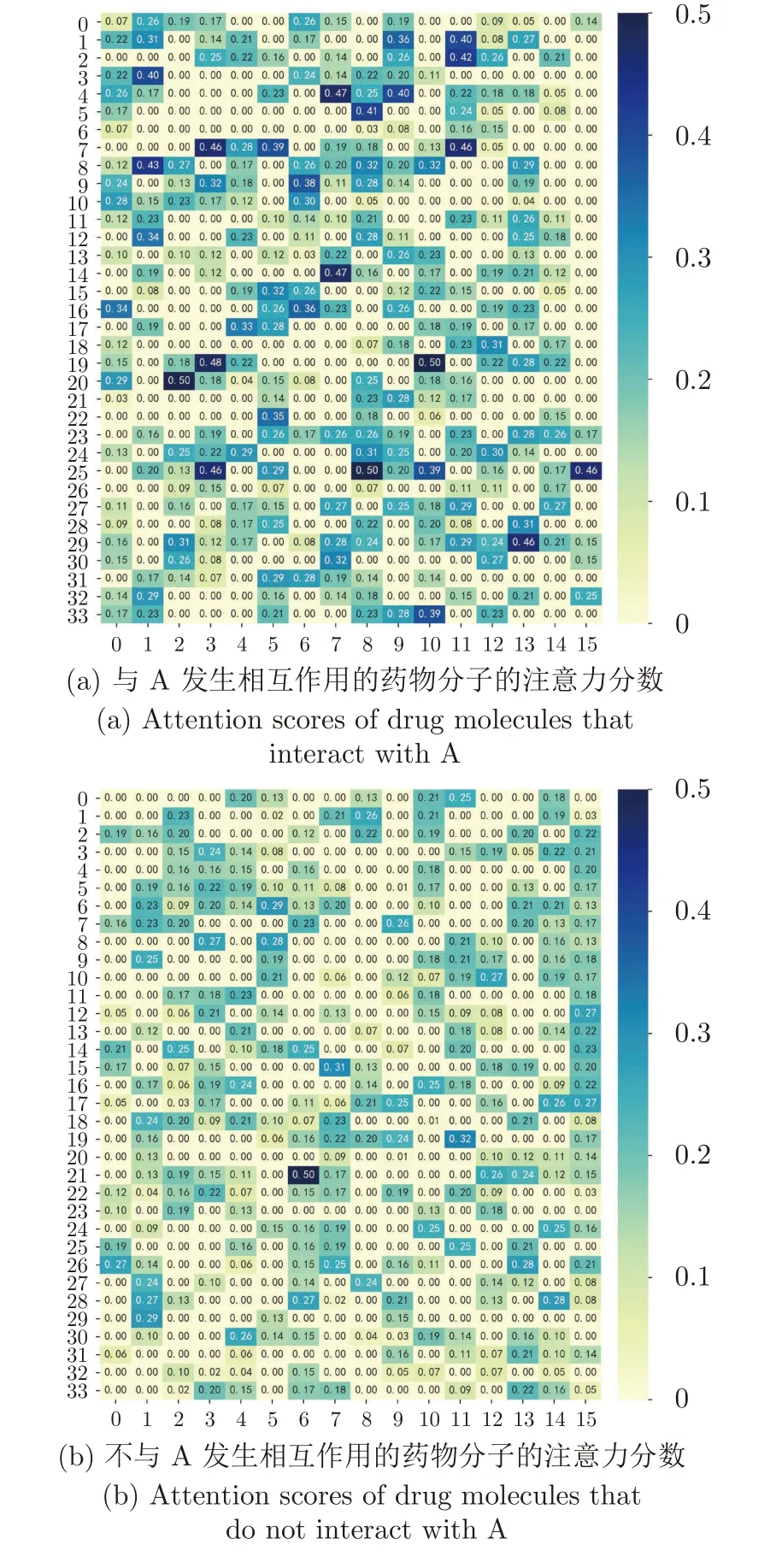
图3 药物分子之间注意力分数的可视化Fig.3 Visualization of attention scores between drug molecules
3.3.2 位置编码的消融实验
为了验证本文提出的基于分子质心的位置编码方法的有效性,本节将研究在有无位置编码和传统位置编码条件下模型性能的差别.表5 是在两个数据集上,本文方法在有无基于分子质心的位置编码和传统位置编码条件下的实验结果.可以看到,本文提出的位置编码方法可以显著提高模型的性能,且使得模型具有更稳定的性能表现.
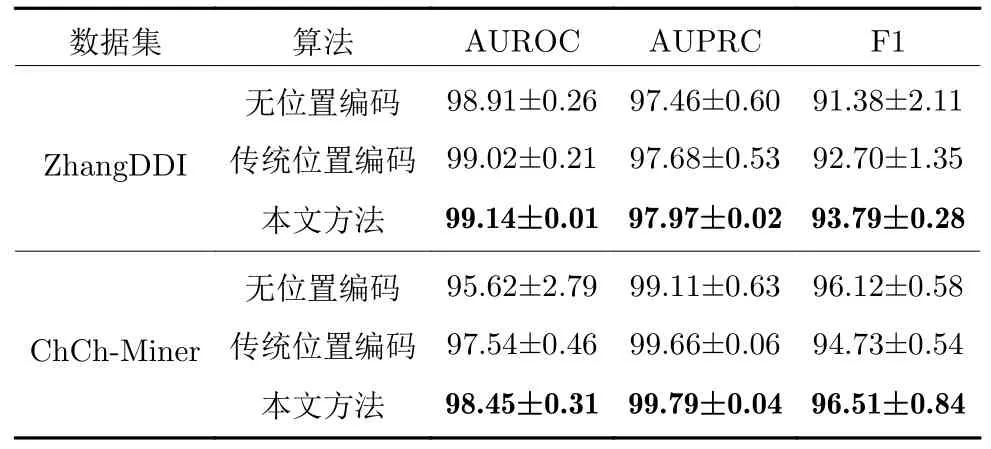
表5 位置编码对模型性能影响的对比结果Table 5 Comparison results of the impact of positional encoding on model performance
如果删去本文提出的位置编码,在原子特征网络学习过程中,将有可能丢失原子节点在药物分子图结构中的相对位置信息;而采用传统位置编码,将使模型仅依赖于各原子节点在SMILES 序列表示中出现的先后顺序,进行原子特征学习.正如前面提到,SMILES 序列是分子的一维线性化表示,因此仅依赖SMILES 序列中原子的先后次序进行原子特征学习,无法充分学到分子的图结构特征信息.
此外,本文提出的位置编码也有助于提高模型的收敛速度.图4 是本文方法在有无位置编码条件下模型性能的收敛曲线,可以清楚地看到,本文提出的位置编码可以显著提高模型的收敛速度.
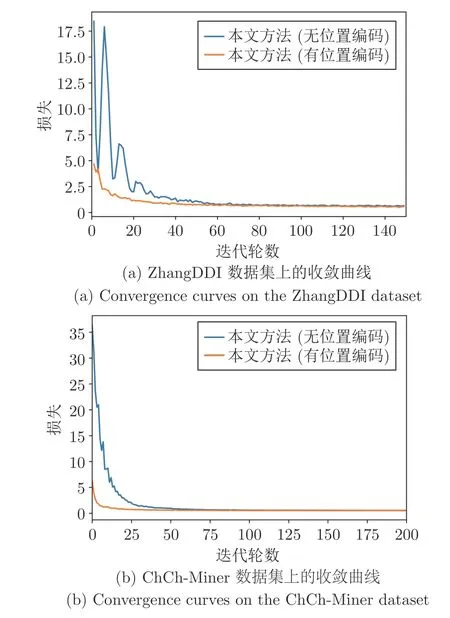
图4 位置编码对模型收敛性能的影响Fig.4 The effect of positional encoding on model convergence performance
与无位置编码的模型相比,本文方法可以在更少的迭代轮数条件下取得更快更好的模型性能.综合表5 和图4 可知,实验结果从侧面进一步证明:基于分子质心的位置编码可以显著提高药物分子中不同原子的编码效率,进而提高模型的收敛速度;同时,通过分子质心引入药物分子的空间结构信息,有助于模型提取更丰富的药物分子结构特征,从而进一步提升模型的DDI 预测精度.
综合上述实验结果可知,本文提出的多层次注意力机制和基于分子质心的位置编码方法都是有效且不可或缺,有助于提高药物分子中不同原子的编码效率和不同药物分子相互作用预测的精度.
3.3.3 损失函数的消融实验
为验证本文引入的自蒸馏约束项和对比学习损失项的有效性,我们在两个数据集上分别针对2 项损失函数进行消融实验,并进一步将对比学习损失项替换为基于互信息的噪声对比估计模型(Mutual information noise contrastive estimation,infoNCE)[22],检验不同对比损失函数对模型性能的影响.此外,我们还改变正负样本采样方式,检验采样方式对本文对比学习损失函数的影响.对于每个药物分子,选取其在分子特征网络的输出特征作为锚点.我们这里选择2 种正负样本的采样方式,其中一种是将锚点的一阶邻居和非一阶邻居在原子特征网络的输出特征分别作为正样本和负样本,即本文实验采用的方法;另外一种是将锚点的二阶邻居在原子特征网络的输出特征作为正样本,其他节点作为负样本,进行对比学习.
表6 是不同损失函数对模型性能影响的对比实验结果.可以看到,无论对于ZhangDDI 还是ChCh-Miner 数据集,在没有基于自蒸馏的正则化约束项或基于对比学习的损失项时,模型性能都有一定程度的降低,即这2 项损失函数对提高模型的性能都不可或缺.对于2 种正负样本采样方式来说,实验结果表明本文选取的采样方式更好.这个结果也表明正负样本采样方式对模型性能具有一定的影响.对于不同对比学习损失函数来说,在ChCh-Miner数据集上,本文方法与infoNCE 相比,取得次优结果;但是在ZhangDDI 数据集上,本文方法取得最优结果,且相对更稳定.这是因为infoNCE 通过自归一化重要性采样来优化互信息的下界,需要相对较多的负样本;而本文采用的JSD 方法则对负样本数相对不敏感,性能也相对更稳定.由此可见,不同对比学习损失函数对模型性能具有一定的影响,本文采用的对比学习损失函数具有一定优势.
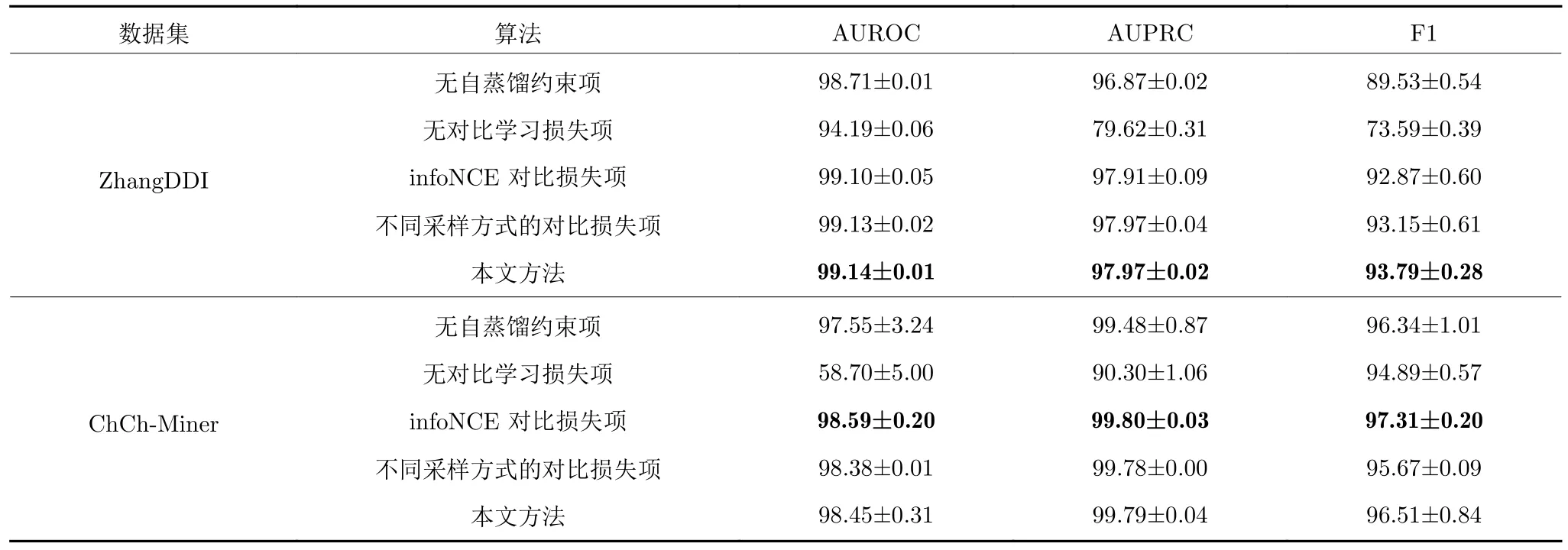
表6 损失函数对模型性能影响的对比结果Table 6 Comparison results of the impact of loss function on model performance
3.4 参数敏感性分析
考虑到损失函数会直接影响模型的性能,本节选择对损失函数中的参数α和β进行敏感性分析.在上述实验中,α和β分别取值1 和0.8.为分析2个参数的敏感性,α取值范围为 {0.2,0.4,0.6,0.8,1},β取值范围为 {0.2,0.4,0.6,0.8,1}.当分析α的敏感性时,β固定,取值为0.8;当分析β的敏感性时,α固定,取值为1.图5 和图6 分别是2 个参数在两个数据集上的实验结果.可以看到,在两个数据集上,不同参数α和β的取值对于本文方法在指标AUROC 和AUPRC 上的结果影响相对较小,而在F1 指标上的结果影响相对较大.综合α和β对模型性能的敏感性分析可知,在α和β分别取值1 和0.8 时,本文方法可以取得最好的实验结果.
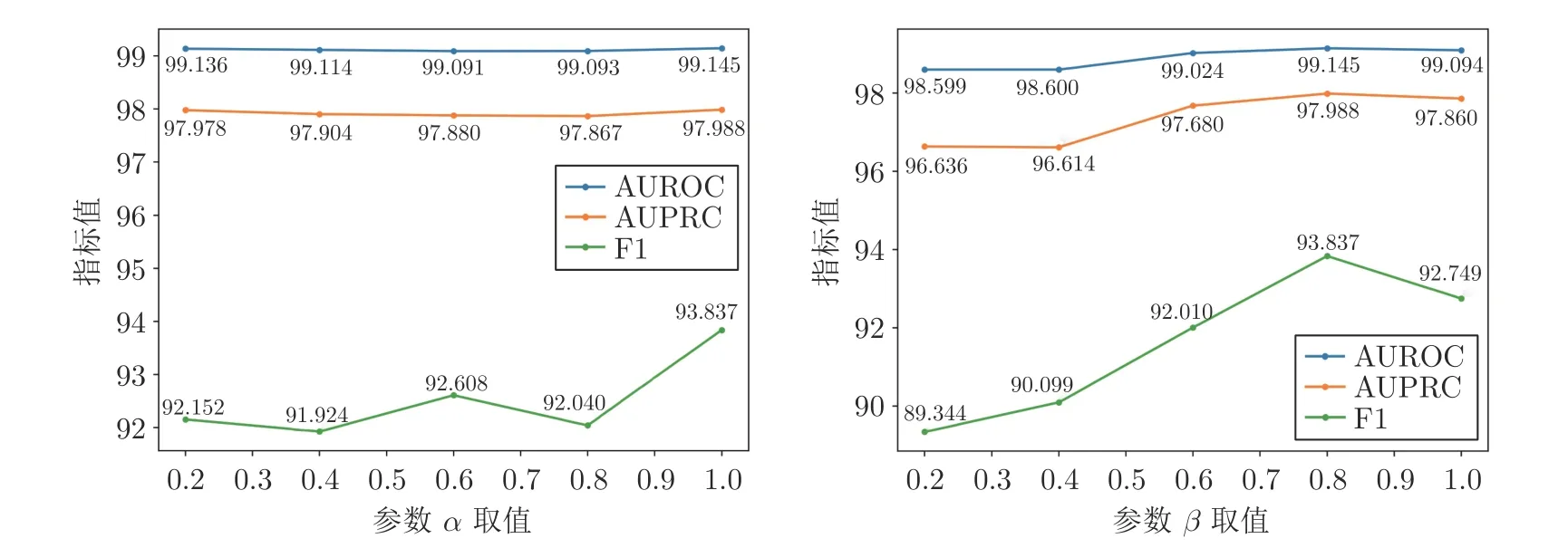
图5 在ZhangDDI 数据集上不同 α 和 β 取值对模型性能的影响Fig.5 The effects of different α and β on model performance on ZhangDDI dataset
4 总结与展望
针对药物相互作用预测的应用研究需求和不同药物分子及其内部不同原子对DDI 预测结果具有不同作用等问题,本文提出一种基于多层次注意力机制和消息传递神经网络的药物相互作用预测方法.通过设计基于注意力机制的原子特征网络和分子特征网络,从两个不同层次分别学习分子内不同原子和化学键以及不同分子间的特征信息,并结合本文提出的基于分子质心的位置编码,提高药物分子编码的效率,从而提高DDI 预测结果的准确性.通过大量对比实验和消融实验验证了本文方法的有效性和优越性.
本文提出的方法虽然可以从原子和分子层面提取药物分子特征信息,但是利用的仅仅只是包含药物分子信息的SMILES 序列,并没有充分利用其他的药物相关信息.下一步的研究工作可以考虑如何充分利用药物分子结构式之外的信息,如同时利用药物分子结构式和包含药物相互作用关系的文本等多种信息,进一步提高模型预测潜在药物相互作用关系的能力.
