类铝离子钟跃迁能级的超精细结构常数和朗德g 因子*
2023-12-01王霞贾方石姚科颜君李冀光吴勇3王建国
王霞 贾方石 姚科 颜君 李冀光† 吴勇3) 王建国
1) (北京应用物理与计算数学研究所,计算物理重点实验室,北京 100088)
2) (复旦大学,核物理与离子束应用教育部重点实验室,上海 200433)
3) (北京大学工学院,应用物理与技术研究中心,北京 100871)
本文利用多组态Dirac-Hartree-Fock 方法计算了类铝等电子序列从Si+到Kr23+离子基组态3s23p 2P1/2,3/2能级的超精细结构常数和朗德g 因子.通过系统评估电子关联效应对Si+和Co14+离子中所关心原子参数的影响,尤其是与内壳层电子相关的关联效应,构建了可靠精确的计算模型,除Si+离子外,超精细结构常数和g 因子的计算误差分别控制在1%左右和10—5 的量级.此外,进一步分析了超精细结构常数中电子部分矩阵元和g 因子随原子序数Z 的变化规律,并拟合了这些物理量与Z 的定量依赖关系,利用拟合公式可以快速计算类铝离子在14 ≤ Z ≤ 54 区间内任意同位素的超精细结构常数和g 因子.
1 引言
高精度的原子光钟既可以作为时间计量基准,也可以检验物理学基本规律或寻找超越标准模型的新物理[1].目前原子光钟的精度已经达到10—19水平[2,3],但是物理学家仍然在思考如何超越现有的精度极限.高电荷态离子(highly charged ion,HCI)中束缚电子受较强的原子核库仑相互作用,不易受环境电磁场的干扰,成为下一代原子光钟的优选对象之一[4—6].理论预言还发现部分HCI 离子钟跃迁频率对精细结构常数随时间的变化比现有原子钟更加灵敏[7],可以用于寻找超越标准模型的新物理.Yudin 等[8]提出类铝离子基组态2P1/2→2P3/2磁偶极跃迁可作为钟跃迁频率标准的候选之一,该体系的优势在于简单的能级结构和极小的电四极偏移.随后,Yu 和Sahoo[9]采用相对论耦合簇(coupled-cluster method with singles and doubles excitation approximation,CCSD)方法系统计算了类铝等电子序列从V10+到Cu16+离子3s23p2P1/2,3/2能级的超精细结构常数和g因子及相关HCI 光钟需要的原子参数,但未考虑与内壳层电子相关的三激发和四激发等高阶关联对相关原子参数的贡献.超精细结构常数对电子关联效应非常敏感,原子态波函数的品质很大程度地影响了计算精度.因此,为了给今后HCI 光钟提供精确可靠的原子参数,有必要分析未被考虑的关联效应对相关原子参数的影响.
此外,原子超精细结构的实验测量与超精细结构常数的精密计算相互结合是确定原子核电磁多极矩的有力工具,通过这种方法确定的原子核电磁多极矩独立于原子核模型,为检验原子核结构理论提供了重要依据[10,11].与中性原子相比,HCI 中原子核与核外电子之间的库仑相互作用占主导,有利于提升原子结构计算精度,获得更可信的原子核性质.类铝离子超精细结构常数的高精度计算将为获取相关核性质提供必不可少的原子参数.
类铝离子的高精度光谱在天体物理和聚变等离子体领域也有重要的应用[12].例如,类铝的Fe13+离子2P1/2→2P3/2跃迁是太阳日冕中可见光范围内的主要谱线之一,并且在其他恒星以及星系星团中也已经观测到这些谱线[13],利用这些谱线可以确定恒星温度和密度等状态参数,为分析恒星形成机制和演化过程提供了理论基础.朗德g因子和超精细结构常数是分析天体物理中磁场强度以及精确测定恒星元素丰度的重要参数[14—16].高精度的类铝离子超精细结构常数和g因子将为今后开展相关光谱分析奠定重要的理论基础.
本文用多组态Dirac-Hartree-Fock (multi-configuration Dirac-Hartree-Fock,MCDHF)方法研究了类铝等电子序列从Si+到Kr23+离子基组态3s23p2P1/2,3/2能级的超精细结构常数和g因子.通过系统分析电子关联效应对Si+与Co14+离子计算结果的影响,发现与内壳层2p 轨道相关的电子关联较为重要,其对超精细结构常数的贡献最大能达到18%.在此基础上,考虑了Breit 相互作用和量子电动力学修正,除Si+离子外,超精细结构常数和g因子的计算精度分别达到1%和10—5量级.本文进一步分析了超精细结构常数中电子部分矩阵元和g因子沿类铝等电子序列的变化规律,并给出了这些物理量与原子序数Z的依赖关系.基于拟合公式计算的超精细结构常数与从头计算的结果符合得很好,因此可以用于计算类铝离子在区间14 ≤Z≤ 54 内任意同位素的超精细结构常数和g因子.
2 理论方法
2.1 MCDHF 方法
在MCDHF 理论框架[17]下,原子态波函数(atomic state wavefunction,ASF)Ψ可由具有相同宇称P和总角动量J及其在z方向分量MJ的组态波函数(configuration state wavefunction,CSF)Φ 线性展开,即
其中,γr和Γ 分别是描述CSF 和ASF 必要的其余量子数,NCSF是组态波函数的个数,cr是组态混合系数.组态波函数是单电子Dirac 轨道波函数构成的Slater 行列式波函数的线性组合.理论上,当组态波函数的个数为无穷多时才能描述真实的原子态波函数,但是在实际计算中只能选择有限个组态波函数,所以组态波函数的选取方式和数目多少决定了电子关联效应的考虑程度,直接影响原子态波函数的品质和原子参数的精度.
基于变分原理,利用自洽场(self-consistent field,SCF)方法同时优化轨道波函数和组态混合系数使所研究的原子态能量达到最低,从而获得一套轨道基组.在获得单电子轨道基之后,利用相对论组态相互作用(relativistic configuration interaction,RCI)方法可以系统地考虑电子关联效应.需要注意的是,RCI 计算过程中只会改变组态混合系数,单电子轨道波函数不再发生变化.另外,在RCI中还可以包括Breit 相互作用和量子电动力学(quantum electrodynamics,QED)修正[18].
2.2 超精细结构常数
超精细相互作用是由原子核的电磁多极矩与电子部分的相互作用引起的[19],相应的哈密顿量表示为
其中T(k)和M(k)分别是电子和原子核部分的k阶张量算符.k=1 表示磁偶极相互作用,k=2 表示电四极相互作用,文中只包括了这两项.磁偶极超精细结构常数为
电四极超精细结构常数为
其中I,μ,Q分别代表原子核的核自旋、磁偶极矩、电四极矩.在原子单位下,电子部分的张量算符T(1)和T(2)分别为
其中,i 是虚数单位;α 是精细结构常数;αj是狄拉克矩阵;rj是第j个电子到原子核的距离;l是轨道角动量算符;C(k)是k阶球张量算符,它的分量与电子部分球谐函数的关系为
从(3)式和(4)式可以看出,超精细结构常数中电子部分矩阵元Ael=AI/μ和Bel=B/Q与原子核无关,其值完全可以由原子结构的计算确定.而且,在精确确定这些矩阵元的基础上,可以从实验上测量的超精细结构中提取相应的核结构参数.
2.3 朗德g 因子
塞曼效应是由原子磁矩和外静磁场之间的相互作用引起的[20],其哈密顿量可写为
其中,μB是玻尔磁子,B为磁感应强度,J为电子的总角动量算符,g为朗德g因子.进一步朗德g因子定义为
其角向部分与超精细相互作用电子部分算符(5)式相似.朗德g因子的QED 修正为
其中gs=2.00232.算符ΔN(1)为
其中,β 和Σ 分别是狄拉克矩阵和相对论自旋矩阵.
3 计算模型
本文计算是利用基于MCDHF 方法发展的GRASP2018 程序包[21].在MCDHF 理论框架下,本文采用活动空间方法系统考虑电子关联效应.根据微扰理论,电子关联可分为一阶关联和高阶关联.一阶关联用单参考组态单双激发形成的组态空间描述,又可进一步划分为价电子之间的关联(valence-valence,VV)、原子芯电子与价电子之间的关联(core-valence,CV)以及原子芯电子之间的关联(core-core,CC).待一阶关联描述充分后,将其组态空间中权重较大的组态加入参考组态基组,用多参考组态基组单双激发形成的组态空间描述髙阶电子关联效应[10,11,22,23].因为从多参考组态单、双激发就相当于从单参考组态限制性的三、四激发,实质上优先考虑了较重要的高阶电子关联效应,所以在有限的计算资源下大大提高了计算效率.在本文的计算中,将3s 和3p 轨道看作价轨道,其余1s,2s 和2p 轨道均看作原子芯轨道.
3.1 VV 和CV 关联
首先,在Dirac-Hartree-Fock (DHF)近似下对参考组态1s22s22p63s23p 的所有占据轨道同时进行优化,并在后续计算中保持不变.随后考虑VV和CV 关联,这里的组态空间通过从参考组态中的占据轨道限制性地激发一个或两个电子到关联轨道产生的.限制条件为原子芯轨道上至多允许一个电子被激发.表1 给出了SCF 计算过程中具体的计算模型以及Si+和Co14+离子2P1/2→2P3/2激发能随组态空间扩大的收敛情况.NCSF列数字代表在每个计算模型下组态的数目.通过计算检验发现h和i等部分高角动量的关联轨道对超精细结构常数和g因子的影响分别小于0.1%和10—8,因此没有将这些轨道加入到活动空间中.如表1 中所列,逐层打开参考组态中占据轨道(active orbitals,AO)的同时,关联轨道(virtual orbitals,VO)也是逐层添加的,并且每次只对新添加的轨道进行优化,把这部分模型标记为VV+CV-n,n代表活动空间的扩展层数.为了考虑更多电子关联时能够达到稳定收敛,活动空间中共包含了10 层关联轨道,其中前2 层轨道角动量包括s,p,d,f 和g,中间2层轨道角动量包括s,p,d 和f,后6 层轨道角动量包括s,p 和d.相比DHF 计算结果,考虑VV+CV模型后,Si+离子激发能几乎没有改变,Co14+离子激发能降低了345 cm—1.

表1 Si+和Co14+离子3s23p 2P1/2,3/2 能级的激发能ΔE (cm—1)随组态空间扩大的收敛趋势.DHF 代表单组态计算模型.AO 和VO 分别代表在每个计算模型下允许被激发的占据轨道和新添加的关联轨道.NCSF 代表相应的组态波函数数目Table 1.Excitation energies ΔE (in cm—1) of 3s23p 2P1/2,3/2 states of Si+ and Co14+ ions as functions of various computational models.DHF stands for the single configuration approximation model.AO and VO represent the occupied orbitals allowed to be replaced and the added virtual orbitals in each computational model,respectively.NCSF represents the corresponding numbers of CSFs.
表2 给出了SCF 计算过程中超精细结构常数电子部分矩阵元Ael和Bel以及朗德g因子随组态空间扩大的收敛情况.可以看出,Ael和Bel呈现不同程度的振荡收敛趋势,g因子最终收敛到10—6.与DHF 计算结果比较,VV+CV 关联对Si+离子Ael和Bel的贡献达到15%以上,而对Co14+离子Ael和Bel的贡献也在7%左右,这表明一阶VV 和CV关联对超精细结构常数计算的重要性.另外,当添加到第9 层和第10 层关联轨道后,Si+和Co14+离子激发能只有0.001%和0.00035%的差异,但是超精细结构常数却相差0.1%和0.04%,这表明超精细结构常数受电子关联的影响更大.
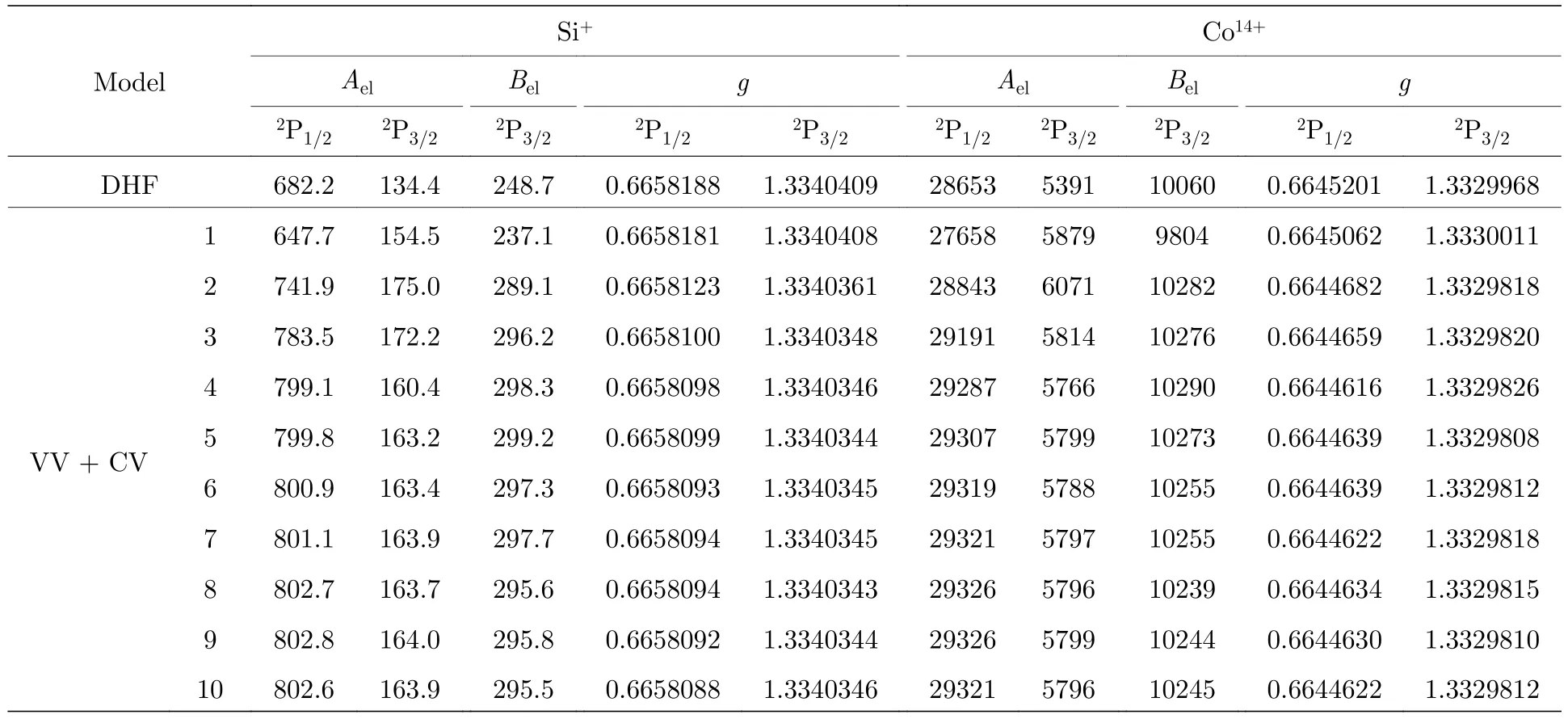
表2 Si+和Co14+离子3s23p 2P1/2,3/2 能级超精细结构常数电子部分矩阵元Ael (MHz/μN)和Bel (MHz/b)以及朗德g 因子随组态空间扩展的收敛情况.DHF 为单组态近似模型Table 2.Electronic parts of hyperfine structure constants Ael (MHz/μN) and Bel (MHz/b) and Landé g factors of 3s23p 2P1/2,3/2 states in Si+ and Co14+ ions as functions of various computational models.DHF stands for the single configuration approximation model.
3.2 CC 关联和高阶关联
在充分描述一阶VV+CV 关联的基础上,采用RCI 方法进一步考虑价壳层轨道的高阶关联以及与内壳层2p 电子相关的CC 和高阶关联,并分析其对计算结果的贡献.表3 给出了不同计算模型下Si+与Co14+离子3s23p2P1/2,3/2能级的激发能、超精细结构常数电子部分矩阵元Ael和Bel以及朗德g因子.同时为了比较,表3 也列出了CCSD 理论的计算结果[9]和NIST 数据库的推荐值[24].从表3 中可以看出,电子关联以及Breit 相互作用和QED 修正对所研究朗德g因子影响较小,因此将系统讨论不同计算模型对激发能和超精细结构常数中电子部分矩阵元Ael和Bel的贡献.
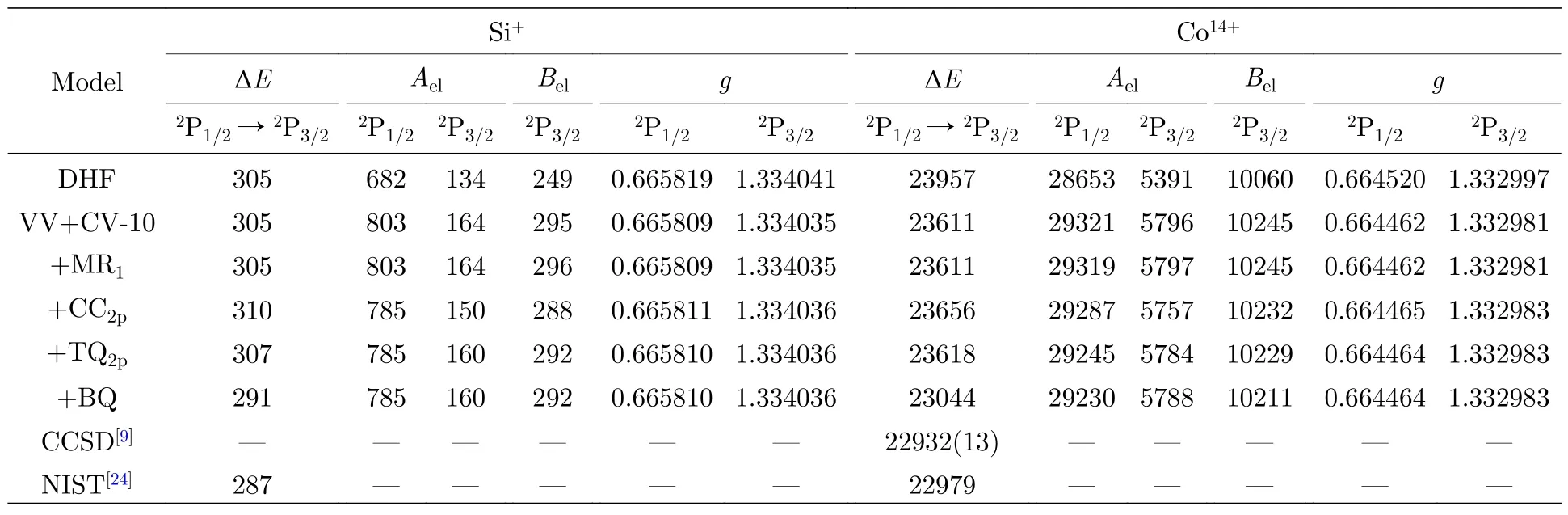
表3 不同计算模型下Si+与Co14+离子3s23p 2P1/2,3/2 能级的激发能ΔE (cm—1)、超精细结构常数电子部分矩阵元Ael(MHz/μN)和Bel (MHz/b)以及朗德g 因子Table 3.Excitation energies ΔE (cm—1),electronic parts of hyperfine structure constants Ael (MHz/μN) and Bel (MHz/b)and Landé g factors of 3s23p 2P1/2,3/2 states in Si+ and Co14+ ions as functions of various computational models.
首先,在VV+CV-10 模型的基础上,考虑价壳层轨道的高阶关联,即从多参考组态(multireference,MR)占据的3s 和3p 电子单双激发到关联轨道所形成的组态空间来描述,标记为MR1.在此模型中,MR 来源于一阶VV+CV 关联原子态波函数中权重大于0.05 的组态,即3p3和3s3p3d;通过监测原子参数随关联轨道扩展的收敛趋势,最终包括5 层关联轨道.考虑MR1模型后,发现Si+和Co14+离子激发能的变化在1 cm—1以内,该模型对Ael和Bel的贡献也不超过0.33%.
在MR1模型的基础上,原子芯中2p 电子的CC 关联可以通过由单参考组态中的2p 电子双激发到所有关联轨道形成的组态来描述,标记为CC2p.研究发现,CC2p模型使Si+和Co14+离子基组态精细结构分裂分别增加了约5 cm—1和45 cm—1(即1.4%和0.2%),对这两种离子Ael和Bel的贡献分别为10%和3%左右.
与2p 电子相关的高阶关联效应是计算中的难点,主要原因是内壳层电子关联效应较弱,在一阶关联原子态波函数中很难挑选出重要的CSFs 作为多参考组态基,因此多参考组态单双激发的方法并不适合考虑与内壳层电子相关的高阶关联效应.本文采用单参考组态限制性三四激发的方式来考虑这部分电子关联,该计算模型标记为TQ2p.通过小规模验证性计算发现f等更高角动量和轨道能较高的关联轨道以及从参考组态占据的2p 轨道上同时激发出4 个电子对Ael和Bel的影响几乎可以忽略不计.因此,TQ2p模型共包含了三层轨道角动量为s,p 和d 的关联轨道;激发方式中允许从2p 轨道和3s,3p 价壳层同时激发1,2,3 和4 个电子,但限制了从2p 轨道的最大激发数到3.TQ2p模型使Si+和Co14+离子基组态精细结构分裂分别减小了约2 和38 cm—1(即0.7%和0.16%),对Ael和Bel的最大贡献可达6%.CC2p和TQ2p模型不仅存在部分相互抵消,对Si+与Co14+离子超精细结构常数的总贡献仍有3.7%和0.3%左右,充分说明这两种电子关联效应对原子参数计算精度的影响不能被忽略.
从两个体系不同计算模型的结果比较可以看出,电子关联效应对原子参数的影响在近中性体系中更显著,且随着原子序数的增加而减小.在以上模型产生的组态空间基础上,对计算结果进行了Breit 相互作用和QED 效应的修正,标记为BQ.这部分修正使Si+与Co14+离子激发能分别降低了16 和574 cm—1(即5.5%和2.5%),对Ael或Bel的贡献分别为0.06%和0.17%,这说明了Breit 相互作用和QED 效应的贡献随原子序数增大而增加.与NIST 数据库的推荐值[24]和CCSD 方法计算的结果[9]相比,激发能的最大差别不到100 cm—1,由此验证了当前计算模型的可靠性,为进一步研究类铝等电子序列超精细结构常数和g因子奠定了一定的基础.
3.3 计算误差
在TQ2p模型的基础上,进一步评估了内壳层2s 和1s 电子的CC 关联以及与其相关的高阶关联对所研究原子参数的影响.组态空间包括了从单参考组态中2s 和1s 轨道同时激发两个电子到所有关联轨道以及限制性三、四激发到部分关联轨道形成的组态波函数.这里对三、四激发的限制条件为至多1 个或2 个电子从2s 或1s 轨道上被激发.另外,高角动量以及轨道能较高的关联轨道对所研究的物理量影响很小,因此生成高阶关联波函数时只包含了三层轨道角动量为s,p 和d 的关联轨道.计算发现描述与1s 和2s 相关的CC 和高阶关联的组态波函数的数量增长了几倍.但是,除对Si+离子2P3/2能级的磁偶极超精细结构常数的贡献在3%左右以外,对其他超精细结构常数的影响均小于1%,对朗德g因子的影响被控制在10—5的水平.因此,为了对所研究的等电子系列涉及的所有原子体系使用相同的计算模型并保证计算效率,最终的计算模型中并没有考虑与内壳层1s 和2s 电子相关的CC 和高阶关联,而将其贡献作为计算误差考虑.值得强调的是,如3.2 节所述,电子关联效应对原子参数的影响随原子序数增加而减小,结果中所给误差是基于等电子系列中近中性离子的评估给出,因此对于离化度较高的体系此误差应比实际误差大.
4 结果与讨论
4.1 Ael,Bel 和g 因子
表4 列出了利用BQ 模型计算的类铝等电子序列从Si+到Kr23+离子基组态3s23p2P1/2,3/2能级的超精细结构常数电子部分矩阵元Ael,Bel和朗德g因子以及相应的计算误差.为了更直观地反映出这些物理量随原子序数Z的变化规律,进一步在图1 中展示了计算结果,如散点所示.从图1 可以看出,超精细结构常数电子部分矩阵元Ael和Bel随原子序数的增加而增大,g因子随原子序数的增加而减小.从(5)式和(6)式可以看出,磁偶极和电四极超精细相互作用分别与原子态波函数径向半径的平方和三次方成反比,而高离化态离子的波函数径向半径近似与原子序数成反比,从中可以定性地解释超精细结构常数电子部分矩阵元与Z的依赖关系.对于朗德g因子,在非相对论近似下可以通过g=1+[J(J+1)—L(L+1)+S(S+1)]/[2J(J+1)]解析得到,给定角动量量子数其值应与原子序数无关,而图1 中g因子随原子序数的变化呈现Z2的依赖关系,可以推断此变化规律是由相对论效应导致的.
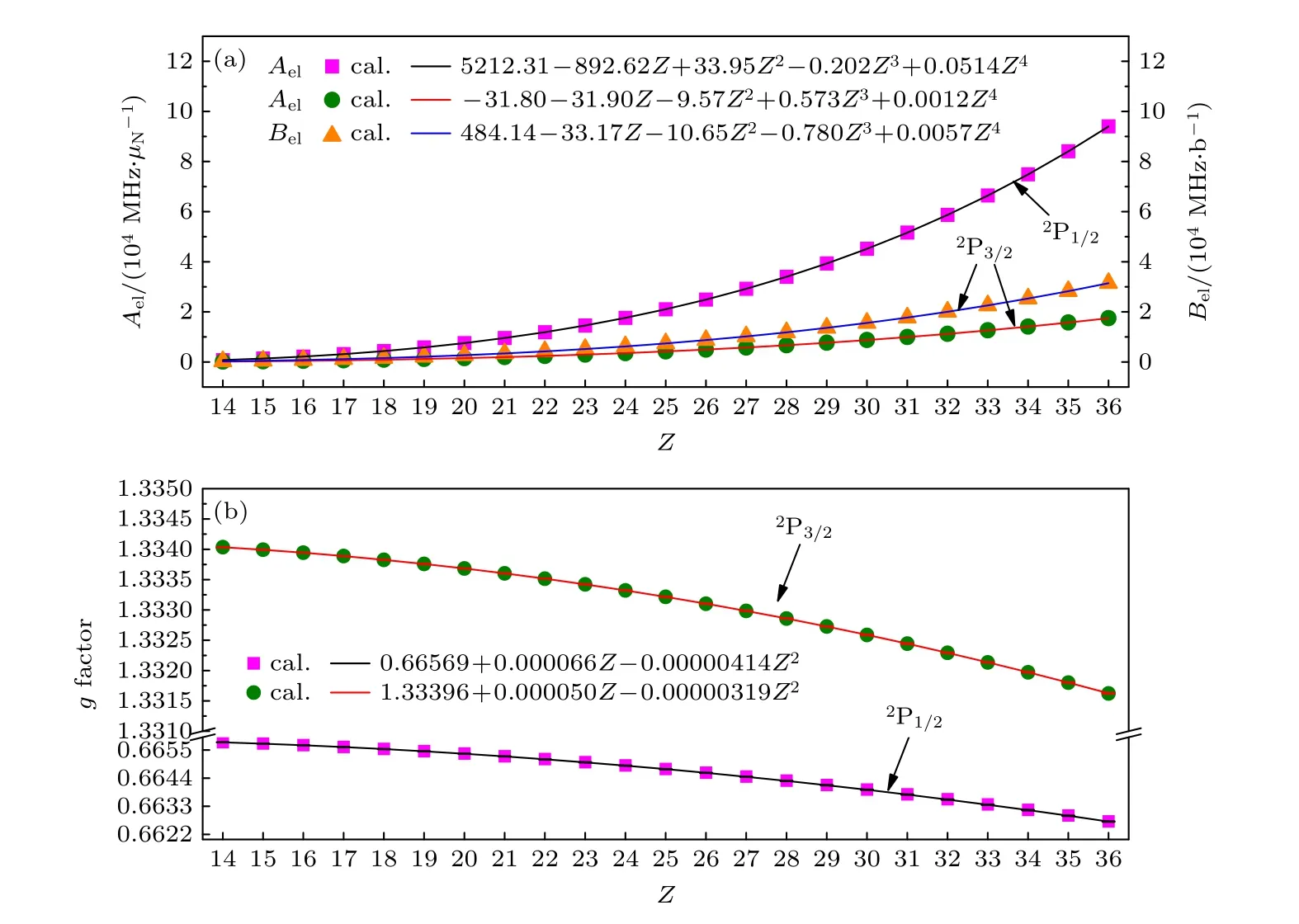
图1 类铝等电子序列3s23p 2P1/2,3/2 能级的(a)超精细结构常数电子部分矩阵元Ael 和Bel 以及(b)朗德g 因子随原子序数Z 的变化关系.图中实线表示由拟合公式得出的结果,散点表示用MCDHF 方法从头计算的结果Fig.1.(a) Electronic parts of hyperfine structure constants and (b) Landé g factors of 3s23p 2P1/2,3/2 states of Al-like isoelectronic sequence ions as functions of atomic number.The solid line represents these results obtained by from numerical fitting formula,and the discrete point represents these results obtained by our ab initio calculation using MCDHF method.

表4 类铝等电子序列3s23p 2P1/2,3/2 能级的超精细结构常数电子部分矩阵元Ael (MHz/μN),Bel (MHz/b)和朗德g 因子.括号内的数字表示计算结果相应的不确定度Table 4.Electronic parts of hyperfine structure constants Ael (MHz/μN) and Bel (MHz/b) and Landé g factors of 3s23p 2P1/2,3/2 states of Al-like isoelectronic sequence ions.Numbers in parentheses represent the computational errors.
进一步,利用多项式拟合了超精细结构常数电子部分矩阵元和g因子与原子序数Z的定量依赖关系,具体表达式列于图1 中.如图1 中实线所示,利用拟合公式计算的Ael,Bel和g因子与从头计算的结果符合得很好,相对偏差小于2%;从头计算的g因子与拟合结果的相对偏差小于10—5.因此,可以利用拟合结果计算此等电子序列区间内任意同位素的超精细结构常数和g因子.同时这些拟合函数可以进一步推广到其他类铝离子,经过理论计算发现,当原子序数Z< 54 时,从头计算和拟合函数结果的相对偏差小于2%,因而该拟合表达式仍然适用,并且对于2P3/2能级的Ael和Bel该拟合表达式还可以推广到更高Z的离子体系.
4.2 A,B 和g 因子
根据稳定同位素的I,μ[25]和Q[26]以及表4 中给出的超精细结构常数电子部分矩阵元Ael和Bel,可以确定类铝等电子序列3s23p2P1/2,3/2能级的磁偶极超精细结构常数A和电四极超精细结构常数B.表5 给出了7 种已建议的下一代HCI 光钟的类铝离子g因子以及稳定同位素的超精细结构常数A和B及相应的计算误差.由表5 可知,除59Co14+离子外,本文计算的A值与CCSD 方法计算的结果[9]差别均小于0.5%,而59Co14+离子两种方法计算的结果相差7 倍以上,原因主要是文献[9]中引用的原子核磁偶极矩有误.两种方法计算的B值差别较大,尤其是对于51V10+,59Co14+离子和55Mn12+,57Fe13+离子,差别分别达到了16%和30%.导致这些差别的原因主要是两种方法采用了不同的原子核电四极矩推荐值[25,26],而电子部分矩阵元计算引起的差别不到0.25%.因此,通过实验测量B值,再结合本文计算的Bel,可以重新确定较为可靠的核四极矩Q.本文计算的g因子与CCSD 计算结果的差别在小数点后第四位或第五位.
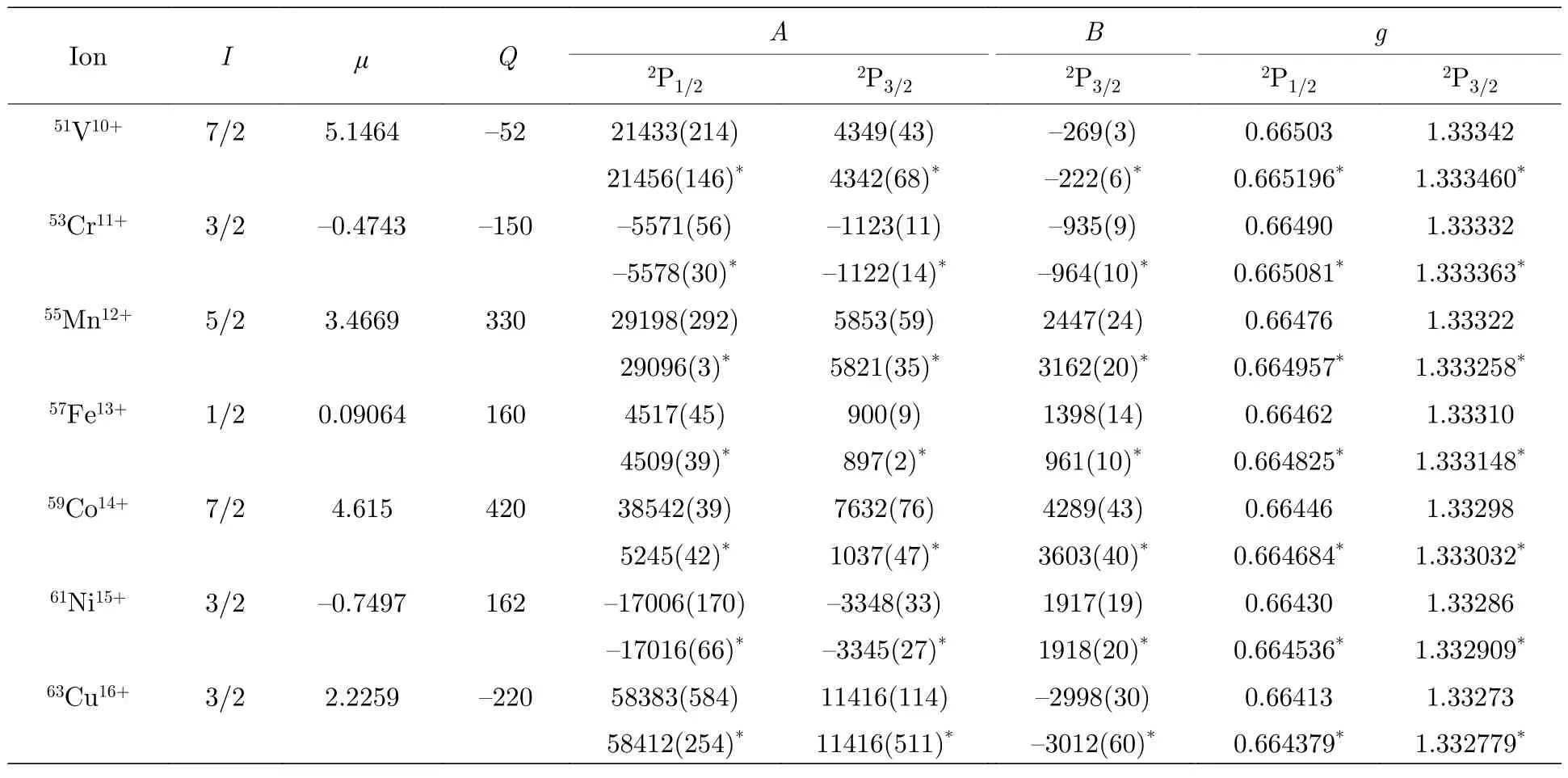
表5 类铝等电子序列3s23p 2P1/2,3/2 能级的超精细结构常数A,B (MHz)和朗德g 因子.所有核参数μ (μN)和Q (mb)均来自于文献[25,26].星号表示用CCSD 方法计算的结果[9].括号内的数字表示计算结果相应的不确定度Table 5.Hyperfine structure constants and g factors of 3s23p 2P1/2,3/2 states of Al-like isoelectronic sequence ions.Nuclear parameters μ (μN) and Q (mb) are taken from Ref.[25,26].Asterisk represents these results calculated by CCSD method[9].Numbers in parentheses represent the computational uncertainties.
5 结论
本文采用MCDHF 方法,研究了类铝等电子序列从Si+到Kr23+离子基组态3s23p2P1/2,3/2能级的超精细结构常数和朗德g因子.通过系统分析了不同类型电子关联效应对Si+与Co14+离子计算结果的影响,构建了可靠精确的计算模型,除Si+离子外,本文计算的超精细结构常数和g因子的精度分别高于1%和10—5水平.进一步,讨论了超精细结构常数和g因子随原子序数的变化规律,并给出了准确的拟合公式,这些拟合函数也可以进一步推广到原子序数小于54 的类铝离子.本文的计算结果为天体物理和原子钟等研究领域提供了可靠的原子参数.
