恩替卡韦的合成
2023-11-03陈宁
陈 宁
(海南建科药业有限公司,海南 海口 571200)
美国医药公司百时美施贵宝(Bristol-Myers Squibb)在1997年以环戊二烯钠为原料经10 步首次成功合成并报道恩替卡韦的全合成路线后[1],2004年10月顺利完成三期临床实验后,2005年被美国FDA批准上市,并于2005年底通过中国食品与药品监督管理局(SFDA)审批在国内上市,由于其“强效低耐药”的特点,投入市场后广受患者好评。
恩替卡韦以博路定为商品名;化学名称为 2-氨基-9-[(1S,3S,4S)4-羟基-3-羟甲基-2-亚甲戊基]-1,9-氢-6-氢-嘌呤-6-酮-水合物;英文化学名为2-amino-1,9-dihydro-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one,monohydrate;分子式为C12H15N5O3·H2O;相对分子质量为295.3;CAS号为 142217-69-4,该分子具有3个手性中心,所以共有8个异构体,而临床上只以上述的(1S,3R,4S)的形式给药。在服药患者体内恩替卡韦由磷酸激酶的作用下形成具有药效的三磷酸化合物形式,拮抗HBV形成所需天然底物脱氧鸟苷三磷酸(GTP),其作用靶点为HBV的逆转录酶,可以在碱基引导(base priming)、mRNA前体反转录成负链以及HBV DNA正链合成[2]这3个环节均抑制HBV逆转录酶活性。该药物在人体内不被肝细胞代谢,主要从肾脏排出体外,在细胞内作用的半衰期为15 h。
恩替卡韦为鸟嘌呤核苷类似物,抗HBV效果强,能够抑制HBV DNA复制的起始、逆转录和DNA正链合成,DNA聚合酶和逆转录酶是HBV作用靶点,经对前基因RNA逆转录复制HBV DNA负链进行抑制,继续对正链合成进行抑制,最终将HBV DNA的装配和延伸阻断[3]。
1 合成方法
通过查阅国内外相关专利文献[4],我们主要确定了三条制备恩替卡韦的合成路线,通过市场调查,我们发现有些工艺用到的原料及试剂价格昂贵,且不易够得,不适合规模化生产。基于化合物专利路线,我们最终确定了合成工艺路线。
(1)合成路线一:参考化合物专利EP481754,申请日:1991.10.16
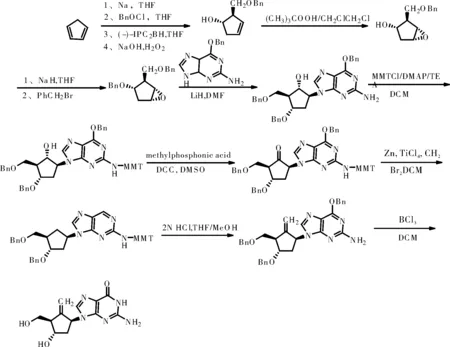
此路线各步收率高,工艺稳定,原料易购得,试剂价格便宜;但是需要用到危险试剂,钠和钠氢,而且多步采用柱层析提纯,不利于工业化生产。
(2)合成路线二:参考文献CN1861602A申请日2005.05.13
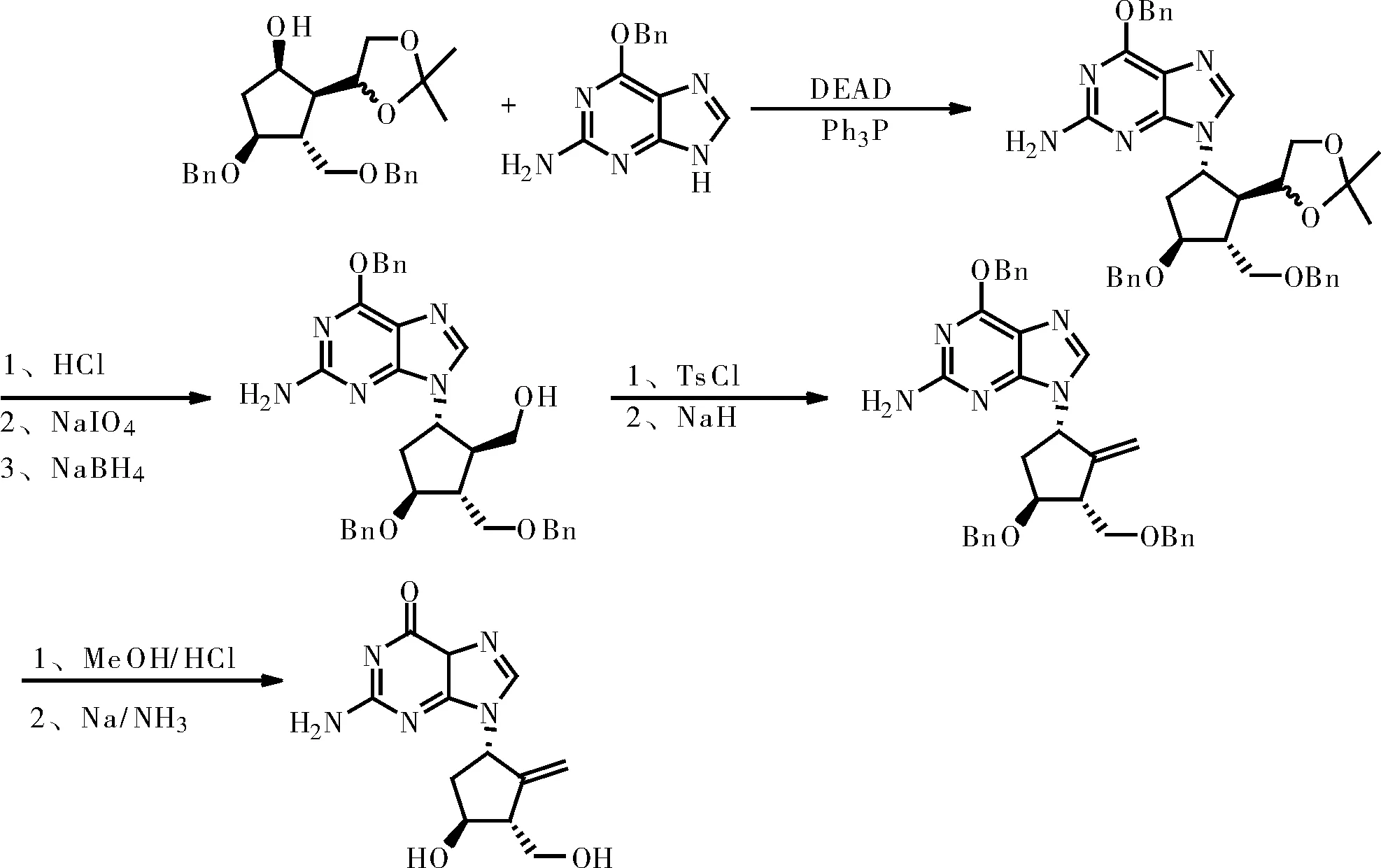
此路线较短,但是原料不易获得,需要用到危险试剂,钠和钠氢。
(3)合成路线三:参考文献CN101531660,申请日2009.04.14
此路线反应较长,反应条件苛刻,且收率较低,不利于工业化生产。
综上所述,我们采用路线一的合成方法,以恩替卡韦中间体5为起始原料合成最终产品恩替卡韦。与其他两条路线相比较,路线一为化合物专利路线,路线长度适中,各步收率高,工艺稳定,原料易购得,试剂价格便宜等优点。我们一中间体5为起始原料避免使用钠、钠氢和锂氢等危险试剂,减少了柱层析等不易操作的纯化过程,有利于工业化生产。
我们优化了工艺,包括:
①中间体N5合成中间体N6:使用了戴斯-马丁试剂做氧化剂,相比化合物专利的氧化剂,戴斯-马丁具有反应条件温和,收率高,后处理方便等优点。反应溶剂也改为易处理的二氯甲烷,反应在超声波下进行,提高反应收率。
②中间体6合成中间体7:直接使用购得的Nysted试剂和四氯化钛体系作为亚甲基话试剂。反应温度范围设定为-60~-70 ℃。
③精制步骤我们采用水重结晶2次的方法,相比化合物专利中采用过树脂提纯的方法,我们的工艺操作简单,适合工业化生产。
该工艺以恩替卡韦中间体N5为起始原料,经过氧化、亚甲基化、水解、脱保护、精制等步骤制得恩替卡韦。
2 实 验
2.1 主要检测试剂和仪器[5]
高效液相色谱仪(色谱柱:用十八烷基硅烷键合硅胶为填充剂(YMC-Pack Pro C18,250 mm×4.6 mm,l.D.S-5 μm,12 nm);流动相:乙腈-水(8∶92);流速1.0 mL·min-1;检测波长254 nm;柱温30 ℃;GC惠普1890II;Bruker ARX-300型核磁共振仪(TMS内标)。
2.2 合成工序
2.2.1 中间体N6的制备
(1)将1.00 kg恩替卡韦中间体N5加入5 L锥形瓶中,加2.0 L无水二氯甲烷溶解待用;
(2)反应瓶放入XC-1200超声波机中,设定频率1000 W,氮气保护下,向反应瓶中加入4.0 L无水二氯甲烷和500 g的DESS-MARTIN试剂,搅拌溶解,控制体系温度在10 ℃以下;
(3)缓慢滴加恩替卡韦中间体N5的3.0 L二氯甲烷溶液,控制滴加速度,体系温度在10 ℃以下,用时约1.5 h;
(4)滴毕,控制反应温度在10 ℃以下,期间用TLC检测反应进度,直至恩替卡韦中间体N5反应完毕,滴完至反应结束约耗时约3 h;
(5)后处理液配制:在100 L处理罐内注入30.0 L的纯化水,搅拌下倒入1.5 kg的硫代硫酸钠及2.0 kg碳酸氢钠,充分搅拌,水浴保持体系温度在10 ℃以下;
(6)后处理:将反应液转入有后处理液的100 L反应釜内,严格控制体系温度在10 ℃以下搅拌30 min后,静置分液;水相用10 ℃以下的二氯甲烷洗涤两次(5 L/次),合并二氯甲烷相,再用10 ℃以下的饱和氯化钠溶液洗涤3次(4 L/次);
(7)有机相保持在10 ℃以下,加入1 kg无水硫酸镁搅拌干燥2 h;
(8)过滤除去干燥剂,10 ℃以下旋蒸至4.0 L左右,待用。
注意事项:严格该控制本步反应操作的温度决不能超过10 ℃。
2.2.2 N7的制备
(1)氮气保护下,加入2.2 L无水THF及5.0 kg的NYSTED试剂,搅拌下加液氮降温至-65 ℃以下;
(2)配制四氯化钛的二氯甲烷溶液:量取875 mL无水二氯甲烷于1 L量筒中,至于数显电子天平上,往其中缓慢加入317.32 g四氯化钛;
(3)保持在-65 ℃以下滴加四氯化钛的二氯甲烷溶液,约60 min滴完,滴加完毕后至少搅拌30 min;
(4)保持在-65 ℃以下滴加恩替卡韦中间体N6-二氯甲烷液,约60 min滴完;
(5)自然升温至-60~-70 ℃反应1 h;
(6)用TLC监测,当原料不再反应时即可终止反应;
(7)后处理液配制:搅拌下,往60 L纯化水内加入3.75 kg碳酸氢钠和18.8 L二氯甲烷,搅拌至少30 min;
(8)将反应液倒入后处理液中,剧烈搅拌1 h后,静置分层;
(9)上层水相用勺子舀出,并用二氯甲烷萃取2次(2.5 L/次),萃取完水相的二氯甲烷相用于滤饼的洗涤。下层有机相用硅藻土助滤,滤饼用二氯甲烷洗涤5次(4 L/次),洗至滤饼显灰白色,二氯甲烷接近无色;
(10)洗涤完的二氯甲烷相同样经由硅藻土助滤,合并滤液,并将滤液静置分层,下层有机相分液得出后,用1 kg无水硫酸镁干燥2 h;
(11)过滤除去干燥剂,浓缩至4 L左右,用4 kg的100~200目硅胶裹料,自然晾干,待过柱分离。
(12)柱层析纯化N7:15 kg的200~300目的大孔树脂装填入玻璃层析柱中,缓慢加入流动相(石油醚∶乙酸乙酯=20∶1)淋洗柱子;
(13)沥干后装入裹好料的待分离样,流动相(石油醚∶乙酸乙酯=20∶1)洗脱;
(14)TLC检测,收集产物部分,旋蒸;
(15)用真空泵抽成泡沫状,以放气泡沫状固体不收缩为终点;
(16)用刮刀捣碎泡沫状固体,平摊到搪瓷托盘中于真空干燥箱30 ℃真空干燥5 h得N7精制品920.01 g。
2.2.3 N8的制备
(1)50 L反应釜中投入10.0 L四氢呋喃,再打开搅拌;
(2)投入中间体7,并注意用1.2 L四氢呋喃将沾壁的N7冲洗干净;
(3)加入计算量的5.5 L 2M盐酸和11.2 L甲醇;
(4)插好温度计,打开水浴循环,设置水浴温度为60 ℃;
(5)体系温度达到50 ℃以后开始计时,保持体系温度搅拌反应3 h;
(6)TLC监测至体系中原料反应完全;
(7)停止加热,反应体系自然降温至室温
(8)3M的碳酸氢钠水溶液中和至中性,继续搅拌析晶12 h;
(9)过滤,滤饼用纯化水共8.0 L洗涤2次后,再用6.0 L乙酸乙酯洗涤2次,于真空干燥箱55 ℃真空干燥5 h,约得425.55 g产物。
2.2.4 恩替卡韦的制备
(1)将装置在带保温设施的反应架上固定后,确保装置在氮气在惰性气体的保护下往反应器内加入425.0 g中间体8及10.0 L无水二氯甲烷;
(2)搅拌,加液氮冷却至-65 ℃;
(3)往其中滴加693.8 g 1 mol/L三氯化硼的二氯甲烷溶液,约1.5 h滴完,滴加过程保持体系温度在-60~-70 ℃;
(4)反应体系升温至-20 ℃左右,保温反应保持3 h;用液氮将体系降温至-65 ℃;
(5)往体系中滴加6.0 L无水甲醇,分1 h滴完,滴加过程保持体系温度在-60~-70 ℃;
(6)反应体系自然升温至室温后转移至旋蒸蒸发仪上旋蒸除去所有溶剂后,加入2 L无水甲醇,再次蒸干,重复2次;
(7)在体系中加入5.5 L纯化水∶甲醇(2∶1)和3.0 L乙酸乙酯,待固体全部溶解,开始分液,乙酸乙酯相用水洗涤2次,合并水相,用6 L乙酸乙酯分3次洗涤;
(8)水相用碱性阴离子树脂pH=7.0~7.5,过滤除树脂,此时析出固体,加热至80 ℃左右溶清自然冷却至5~10 ℃析晶5 h,过滤收集晶体1;
(9)晶体1于5.7 L水∶甲醇(2∶1)中加热至80 ℃溶解,加入活性炭26 g,继续加热回流15 min,趁热过滤至D级洁净区,冷却至5~10 ℃析晶5 h,过滤得到晶体2。真空干燥箱内45 ℃干燥至恒重。得到恩替卡韦-水合物大约185 g。

图1 恩替卡韦的合成
高效液相色谱仪(色谱柱:高效液相色谱仪(色谱柱:用十八烷基硅烷键合硅胶为填充剂(YMC-Pack Pro C18,250 mm×4.6 mm,1.D.S-5 μm,12 nm);流动相:乙腈-水(8∶92);流速1.0 mL·min-1;检测波长254 nm;柱温30 ℃。测试含量99.11%,水分5.9%,总收率55.06%。恩替卡韦结构示意图如图2所示,1H-NMR数据如表1所示。

表1 H-H COSY

图2 恩替卡韦的结构
供试品1H-NMR谱显示出13组氢,共15个质子,其中2对氢峰基本对称,其积分比(由高场到地场)约为1∶1∶1∶2∶1∶1∶1∶1∶1∶1∶2∶1∶1,重水交换图谱显示有5个活泼氢,与恩替卡韦化学结构吻合。
(1)δ2.02-2.06~2.19-2.25(两个多重峰,2H),结合本品DEPT谱和HSQC谱可知,这两个氢峰属于仲碳质子。在H-H COSY图谱中,这两个氢峰呈现相关关系,这组氢峰处于最高场,应分别属于C5’-Ha,C5’-Hb的信号峰。
(2)δ2.5~2.53(多重峰,1H),结合本品DEPT谱和HSQC谱可知,该氢信号属于叔碳质子。化学位移较高,应属于C3’-H的信号峰。
(3)δ3.52~3.55(多重峰,2H),结合本品DEPT谱和HSQC谱可知,该氢属于仲碳质子。在H-H COSY图谱中,该氢信号与δ2.5~2.53(C3’-H)和δ4.84~4.86(活泼氢)的信号相关,应为C7’-2H的氢信号,δ4.84~4.86处的氢信号应为C7’-OH的信号。
(4)δ4.23(单峰,1H),结合本品DEPT谱和HSQC谱可知,这两个氢属于叔碳质子。在H-H COSY图谱中,该信号与δ2.19~2.25(C5’-Hb)和δ4.88~4.89(活泼氢)的信号相关。所以,δ4.23应为C4’-H的氢信号,δ4.88~4.89为C4’-OH的氢信号。
(5)δ4.56~5.10(两个单峰,2H),结合本品DEPT谱和HSQC谱可知,这两个氢峰属于仲碳质子。在H-H COSY图谱中,这两组氢信号与δ2.5-2.53(C3’-H)相关,化学位移较高,应为C6’-Ha和C6’-Hb的氢信号。该质子受sp2杂化碳原子的诱导和中等程度的双键各向异性效应作用,化学位移较大。
(6)δ5.34~5.38(多重峰,1H),结合本品DEPT谱和HSQC谱可知,此氢为叔碳氢。在H-H COSY图谱中,该峰与δ2.02-2.06~2.19-2.25(C5’-Ha,C5’-Hb)的氢信号相关,应为C1’-H的信号峰。该信号由于受到氮原子的诱导效应和芳环的各向异性效应,化学位移高。
(7)δ6.44(单峰,2H)和δ10.63(单峰,1H),这两组信号在重水交换图谱中消失,应为活泼氢,化学位移高的为N1-H的信号峰,δ6.44为C2-NH2信号峰。
(8)δ7.67(单,1H),结合本品DEPT谱和HSQC谱可知,此氢为芳环上的叔碳氢,由于受到芳环屏蔽效应的影响,化学位移处于低场,为C8-H的信号峰。
此解析结果与恩替卡韦结构相符,供试品与对照品氢谱基本一致。
3 结果与讨论
(1)在中间体N6的制备过程中,使用了戴斯-马丁试剂做氧化剂,戴斯-马丁具有反应条件温和,收率高,后处理方便等优点。反应瓶放入XC-1200超声波机中,设定频率1000 W,反应收率高。反应溶剂也改为易处理的二氯甲烷。
(2)中间体6合成中间体7:直接使用购得的Nysted试剂和四氯化钛体系作为亚甲基话试剂。生产要求的反应温度为低于-60 ℃,这一反应条件耗能高,会对成品的成本产生影响,所以筛选反应不同温度,结果发现处于-70 ℃时,反应时间为1 h,收率为64%;处于-60 ℃时,反应时间为1 h,收率为44%;处于0 ℃以下时,反应时间为1 h,收率为2%;处于10 ℃时,反应时间为1 h,收率为0。由此得知,反应温度范围设定为-60~-70 ℃。
(3)采用水∶甲醇(2∶1)重结晶2次的方法,相比化合物专利中采用过树脂提纯的方法,我们的工艺操作简单,适合工业化生产。经检验各项指标均符合2020版药典质量标准。
4 结 论
(1)最佳条件确定为:在中间体N6的制备过程中,使用了戴斯-马丁试剂做氧化剂,反应在超声波频率下进行,设定频率1000 W,反应溶剂也改为易处理的二氯甲烷;中间体6合成中间体7:直接使用购得的Nysted试剂和四氯化钛体系作为亚甲基话试剂,反应温度设定为-60~-70 ℃;采用水∶甲醇(2∶1)重结晶2次的方法,最终得到恩替卡韦成品。
(2)测试含量99.11%,水分5.9%,质量收率55.06%。
(3)通过这次的工艺优化和3批生产验证,表明该工艺适合规模化生产,反应条件可控,经检验各项指标均符合2020版药典质量标准。
