Ni@Silicalite-1催化剂的制备及其催化CO2加氢制CH4与CO性能研究
2023-11-01张嘉兴王明瑞周阿娟党飞雄张光辉张安峰郭新闻
赵 玉,张嘉兴,王明瑞,周阿娟,党飞雄,张光辉,张安峰,郭新闻
(大连理工大学 化工学院 精细化工国家重点实验室,智能材料化工前沿科学中心,辽宁 大连 116024)
近年来,随着化石能源的不断消耗,大量温室气体CO2被排入大气中,引发一系列环境问题。将捕集的CO2与由清洁能源,如光能、风能等生产的“绿氢”在催化剂的作用下进行高效耦合,制备相应的化学品受到了强烈关注[1-2]。其中,Ni基催化剂因其对H2活性能力强、加氢活性高的特点而在CO2加氢转化中的研究较为广泛[3-4]。通过调变Ni 物种的价态组成与结构特性,以控制其加氢能力,进而实现对产物选择性的调控,是目前Ni基催化剂在CO2加氢反应研究中的重要方向[5]。
利用封装策略可增强Ni 物种与载体间的相互作用,优化其催化结构并增强结构稳定性,是目前相关研究的重要方向[6-7]。YANG等[8]利用SSZ-13分子筛的纳米笼封装NiO颗粒,还原后得到了主要暴露(111)晶面的Ni0颗粒,形成的Ni-SSZ-13界面有利于Ni0物种的稳定,催化剂在450 ℃下表现出较高的抗烧结特性与热稳定性。MⅠAO等[9]利用超声辅助,以先封装后浸渍的方法合成了Ni/ZSM-5@MCM-41复合催化剂,该复合催化剂在高CO2转化率(80%)与高CH4选择性(97%)下还表现出较高的稳定性(约80 h)与抗积炭特性,原位漫反射红外(in situ-DRⅠFTS)实验表明了Ni0颗粒上解离活化形成的*H物种参与到表面碳酸氢盐(HCO3-)向甲酸盐(HCOO-)的转化过程中,随后HCOO-进一步加氢分解形成CH4。LⅠU 等[10]利用盘状SBA-15 分子筛与Ni 物种间的强相互作用,构筑更多强吸附CO2与*CO中间体的活性Ni0位点,显著提高了催化活性。此外,HONGMANOROM等[11]合成了具有有序介孔结构的CeO2封装的Ni基催化剂,其在催化CO2加氢甲烷化中的转化频率(TOF)为浸渍型Ni催化剂的3倍。上述工作为探究Ni 催化剂中CO2加氢转化机制与提高催化剂活性及稳定性提供了重要借鉴,然而,由于研究中的活性物种依然局限在Ni0,其加氢产物仍主要为CH4。Silicalite-1(简写为“S-1”)分子筛具有孔道丰富、比表面积高及机械性能好的特点,被广泛用作Ni 基催化剂的载体,以提高金属分散度,增强催化材料的抗烧结、抗积炭性能等[12-13]。通过S-1分子筛封装的策略以调控Ni物种的价态与结构组成,进而实现对CO2加氢产物的选择性合成,是目前研究中的关键课题。
本文通过浸渍-再晶化的方法制备封装型Ni@S-1 催化剂,加强载体与金属的相互作用,提高Ni基催化剂在临氢条件下的稳定性,并讨论封装前后催化剂的Ni 物种的结构演变及其对应的CO2加氢性能,构建Ni活性相组成与加氢选择性间的构效关系,为加氢产物CO 或CH4的选择性生成提供指导与借鉴。
1 实验部分
1.1 实验材料与试剂
六水合硝酸镍(Ni(NO3)2·6H2O):分析纯,天津大茂化学试剂厂;四丙基氢氧化铵(TPAOH,质量分数25%):彩瑞化学试剂有限公司;硅溶胶(质量分数30%):青岛海湾精细化工有限公司;正硅酸乙酯(TEOS):分析纯,西陇化工股份有限公司;无水乙醇(C2H5OH):分析纯,天津市富宇精细化工有限公司;氢氧化钠(NaOH):分析纯,天津市大茂化学试剂厂;硼氢化钠(NaBH4):分析纯,国药集团化学试剂有限公司;间硝基甲苯(C7H7NO2):分析纯,上海麦克林生化科技有限公司。
1.2 催化剂制备
1.2.1 Ni/X催化剂
采用等体积浸渍法合成Ni/X 催化剂(X 为SiO2或S-1 分子筛,下同),采用Stober 法制备Ni/SiO2催化剂中的纳米级硅球[14-15]。称取0.78 g Ni(NO3)2·6H2O 溶于0.96 g 去离子水中,逐滴滴加至2.00 g 硅球粉末中并不断搅拌,浸渍完成后于室温下静置24 h,放入80 ℃烘箱中6 h,在马弗炉空气气氛下540 °C 焙烧6 h,所得到的样品命名为10% Ni/SiO2(10%为Ni/SiO2催化剂中NiO的质量分数)。
采用水热合成法制备Ni/S-1催化剂中的S-1 分子筛。称取0.37 g NaOH 溶解于52.95 g 去离子水中,再加入3.12 g TPAOH 和16.00 g TEOS,在40 ℃下搅拌水解3 h至溶液呈澄清透明状态,装入100 mL水热晶化釜中,在170 ℃烘箱中晶化24 h。晶化结束后取合成产物离心,随后放入80 ℃烘箱中12 h,最后将催化剂放入马弗炉中,在540 ℃下焙烧6 h以去除模板剂,得到的样品标记为S-1。金属Ni负载量(质量分数,下同)为5%和10%的Ni/S-1催化剂制备方法为分别称取0.39 g 和0.78 g 的 Ni(NO3)2·6H2O溶于1.10 g 去离子水中,将溶液分别逐滴滴加至2.00 g S-1 固体粉末中并不断搅拌,浸渍完成后于室温下静置24 h,放入80 °C 烘箱中6 h,最后在马弗炉中540 °C 焙烧6 h,所得到的催化剂分别命名为5% Ni/S-1和10% Ni/S-1。
1.2.2 Ni/X@S-1催化剂
采用再晶化法合成Ni/X@S-1催化剂。Ni/SiO2@S-1催化剂的制备方法为称取2.43 g TPAOH、37.00 g去离子水和6.00 g质量分数30%的硅溶胶,35 °C下搅拌30 min,加入上述制备的10% Ni/SiO2母粉搅拌30 min后装入晶化釜,170 °C、30 r/min旋转晶化24 h。晶化完成后取出离心,随后放入80 °C烘箱中12 h,最后在马弗炉空气气氛下540 °C焙烧6 h,所得到样品命名为Ni/SiO2@S-1(理论负载量为5% NiO)。此外还考察了模板剂用量(n(TPAOH)/n(SiO2) =0.05、0.10)和晶化时间(24 h、72h)对合成效果的影响。Ni/S-1@S-1 催化剂的制备方法为称取2.43 g TPAOH、37.00 g去离子水和6.00 g质量分数30%的硅溶胶,在35 °C 下搅拌30 min,加入上述制备的5% Ni/S-1 或10% Ni/S-1 母粉搅拌30 min 后装入晶化釜,170 °C、30 r/min 下旋转晶化24 h。晶化完成后取出样品离心,随后放入80 °C烘箱中12 h,最后在马弗炉中540 °C 焙烧6 h,所得到的样品命名为2.5% Ni/S-1@S-1与5.0% Ni/S-1@S-1(2.5%和5.0%是指Ni/S-1@S-1催化剂中NiO的质量分数)。
1.3 表征方法
1.3.1 催化剂形貌观测
催化剂的形貌信息与表面特性采用扫描电子显微镜(SEM)进行观测。仪器型号为Hitachi SU8200,加速加压为5 kV。测试前,将粉末样品超声分散于乙醇中,滴加在硅片表面。在表面乙醇全部挥发后,粘附在样品台导电胶带上以备观测。
1.3.2 催化剂元素测试
催化剂元素分布采用场发射扫描透射电子显微镜(STEM)-能量散射型X射线荧光光谱仪(EDX)进行分析,测试电压为200 kV。测试前粉末样品超声分散于乙醇中,再滴加到碳膜载体上。
催化剂元素含量采用美国Perkin EⅠmer公司生产的OPTⅠMA 2000DV型电感耦合等离子体发射光谱(ⅠCP)仪进行测定。
1.3.3 催化剂结构测试
催化剂的比表面积与孔容等结构参数采用美国康塔公司Quadrasorb SⅠ型物理吸附仪上通过N2物理吸附进行测定。使用压片成型样品,首先在300 °C 下脱气处理6 h,再于-196 °C 下进行吸附测试。比表面积采用多点Brunauer-Emmett-Teller(BET)方法进行计算。
不同气氛下(H2-预还原与CO2加氢反应)催化剂的晶体组成和结构采用原位XRD技术测试,所用仪器为Rigaku Smartlab 9 kW X 射线衍射仪,Cu Kα作为靶材,X 射线出射波长为0.154 nm,测试电流、电压分别为45 mA、200 kV,测试范围为10°~60°,扫描速率为8 (°)/min。采用XRK 900 反应池进行原位测试,反应温度由TCU 750 温控模块控制。在预还原阶段通入纯H2气体,反应阶段通入混合气(V(CO2)/V(H2)/V(N2) = 21/63/16),测试压力为0.1 MPa,气体流量为30 mL/min。在测试过程中,在样品池凹槽内装入样品并压平,通入N2完全置换池内残留空气后,通入H2并升温至400 ℃开始预还原。还原阶段结束后,进行1 h N2气体吹扫,并将降至反应温度(300 ℃)后,通入反应气体进行测试。对于未经过预处理的样品在N2吹扫后直接升至300 ℃在CO2反应气氛下进行测试,采用1-dimension检测器收集XRD 数据,并将其与JCPDS 标准卡片进行对比分析,以确定相应的物相组成。每个测试温度点测定至结构保持后至少1 h,仅标注测试温度的数据点表示在该条件下1 h 内谱图形状无明显变化。
1.3.4 大分子液相加氢测试
测试前,将粉末样品加水超声,加入硼氢化钠敞口反应10 min,以将NiO完全还原为Ni0。再加入水和间硝基甲苯于35 °C 水浴搅拌40 min,用针管吸出0.6~0.7 mL 于25 mL 容量瓶定容。采用V570 型分光光度计获得样品的紫外-可见光光谱。
1.3.5 催化剂还原性能测试
采用美国康塔仪器公司的 ChemBET Pulsar 化学吸附仪进行H2-TPR 测试,以明确样品的还原性质。称取约100 mg 样品装于U 型管中,测试前用He在400 °C下吹扫40 min,后降至室温,切换至5%H2/Ar,并以10 °C/min的速率升温至800 °C,通过热导检测器(TCD)检测脱附气体的信号。
1.4 CO2加氢反应性能测试
CO2加氢反应性能采用固定床反应器评价,反应管为内径8 mm 的石英管,称取200 mg 样品与200 mg 石英砂均匀混合后填入管内恒温段。催化剂预处理条件为:分别在320 ℃、380 ℃和400 ℃温度下,H2气氛中预还原2 h,后切换至N2吹扫降温至300 ℃;或不经历预处理过程,直接进行催化反应。反应条件为:300 °C、0.1 MPa,反应气组成为V(CO2)/V(H2)/V(N2) = 21/63/16,总质量空速为20000 mL/(g·h)。反应产物采用配有TCD 和FⅠD 检测器的Agilent 7890B 在线色谱进行检测分析,其中TCD 检测CO2、N2、CH4和CO,FⅠD 检测CH4与其他烃类产物。取样时间为切换至反应气1 h 后。CO2转化率(XCO2,%)、产物选择性(Si,%)与产物生成速率(Ri,mmol/(g·h))分别根据式(1)~式(3)进行计算。
式中,nCO2,in与nCO2,out分别代表入口与出口处的CO2浓度,mol/L;mi代表产物i的碳数;ni,out为出口处产物i的浓度,mol/L;GHSV为总反应空速,20000 mL/(g·h) ;Vm为标准状况下的气体摩尔体积,22.4 L/mol。
2 结果与讨论
2.1 催化剂形貌分析
采用SiO2纳米球作为载体,利用浸渍-再晶化法合成封装型催化剂,SiO2纳米球与不同合成条件的Ni/SiO2@S-1催化剂的SEM照片如图1所示。
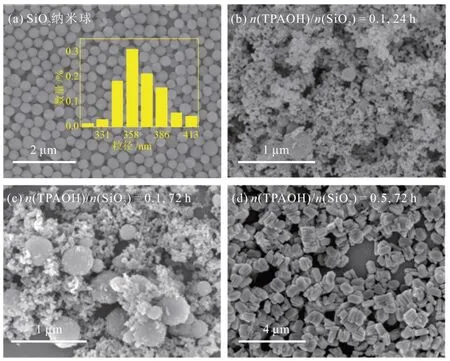
图1 SiO2 纳米球与不同合成条件的Ni/SiO2@S-1 催化剂的SEM照片Fig.1 SEM images of SiO2 nanospheres and Ni/SiO2@S-1 catalysts with different preparation conditions
由图1(a)可知,制备的SiO2纳米球呈规则球状形貌,颗粒尺寸集中在(360 ± 20) nm。SiO2纳米球再晶化过程中不存在模板导向作用,因此模板剂TPAOH 的加入量与晶化时间对再结晶形成S-1 分子筛起到关键作用。由图1(b)和图1(c)可知,在低TPAOH 加入量(n(TPAOH)/n(SiO2) = 0.1)时,延长晶化时间(24~72 h)并未明显促进S-1 分子筛的再晶化,在SEM 照片中仍能观察到较多未被包覆的SiO2纳米球与大量未完全晶化的无定形物质。在较高TPAOH 加入量(n(TPAOH)/n(SiO2) = 0.5),且晶化较长时间(72 h)时,得到的大部分样品颗粒呈现规则的六边形板状形貌的S-1 分子筛,表明此时再结晶形成的S-1 分子筛包覆在SiO2纳米球外,并且结晶性良好。但仍有少量未晶化而形成对应的颗粒,分散在其周围。
不同质量分数Ni 的Ni/S-1 与Ni/S-1@S-1 催化剂的SEM照片见图2。

图2 不同Ni 质量分数的Ni/S-1 与Ni/S-1@S-1 催化剂的SEM照片Fig.2 SEM images of Ni/S-1 and Ni/S-1@S-1 catalysts with different mass fractions of Ni
由图2(a)和图2(b)可知,两者均呈现规整的S-1六边形板状形貌,其中b轴长度约为180 nm,c轴长度约为330 nm。催化剂外表面较为光滑,并未观察到明显的NiO 颗粒,表明焙烧后的NiO 物种依然在S-1分子筛上高度分散。由图2(c)和图2(d)可知,采用S-1 分子筛进行再结晶封装后,催化剂仍保持规则平整的形貌,但分子筛颗粒尺寸明显增加,其中b轴长度增大至220 nm左右,c轴长度增大至450 nm左右,但在其外表面仍未观察到明显金属颗粒的存在。
将5.0% Ni/S-1@S-1 催化剂进行能量散射型X射线荧光光谱仪(EDX)元素分布分析,结果如图3所示,Ni物种的信号主要集中在S-1内部,在包覆层中无明显Ni信号,进一步说明封装效果较好。上述结果表明,在再晶化过程外部形成的S-1 分子筛可以包覆内部的原有沸石与NiO颗粒,进而将Ni物种限域在重结晶的S-1 分子筛的内部,未见有明显的金属浸出或团聚。
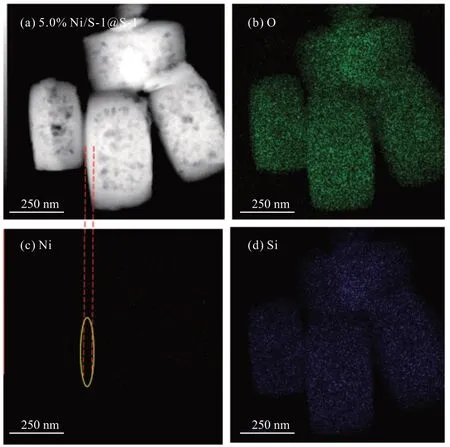
图3 5.0% Ni/S-1@S-1催化剂的EDX元素(Ni、O和Si)分布Fig.3 EDX elements (Ni, O and Si) distribution of 5.0% Ni/S-1@S-1 catalysts
综上所述,相对于SiO2纳米球载体,以S-1为载体时,可充分利用其自身的模板导向作用,促进再晶化过程,更有利于合成形貌规整、晶化完全的封装Ni基催化剂。
2.2 催化剂N2物理吸附分析
负载型的5% Ni/S-1与封装型5.0% Ni/S-1@S-1催化剂的孔结构特性如表1 与图4 所示。S-1 封装后催化剂的比表面积由323 m2/g 增大至418 m2/g,归因于包覆的S-1提供的丰富的孔道结构。由图4(a)可知,5% Ni/S-1与5.0% Ni/S-1@S-1均在p/p0= 0.40出现明显的回滞环,呈现典型的Ⅳ型吸脱附曲线,表明材料孔道存在介孔。 由图4(b)可进一步表明上述材料有介孔的存在(2~50 nm)。

表1 不同催化剂的孔结构特性Table 1 Pore structure properties of different catalysts
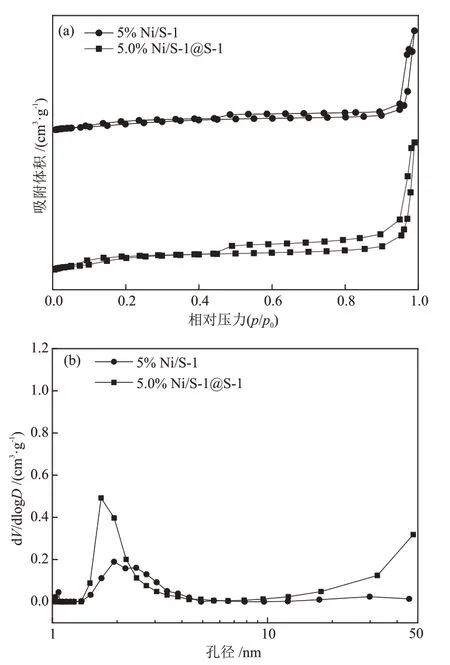
图4 不同催化剂的N2吸/脱附等温线(a)及孔径分布(b)Fig.4 N2 adsorption/desorption isotherm curves (a) and pore size distribution (b) of different catalysts
2.3 催化剂大分子液相加氢及ICP元素分析
为了比较封装前后表面金属含量的变化,分别对后续反应评价所需的5% Ni/S-1及5.0% Ni/S-1@S-1催化剂进行大分子液相加氢测试。大分子液相加氢是通过NaBH4将NiO 还原为Ni0并与间硝基甲苯发生反应生成间胺基甲苯,间硝基甲苯由于分子尺寸较大不能进入微孔孔道,当金属完全封装在内部的时候,则不会发生反应。由图5 可知,5% Ni/S-1催化剂出现明显的峰,说明S-1 表面的单质Ni 与间硝基甲苯发生反应;5.0% Ni/S-1@S-1催化剂未出现峰,说明再晶化过程可以实现NiO的颗粒有效封装。
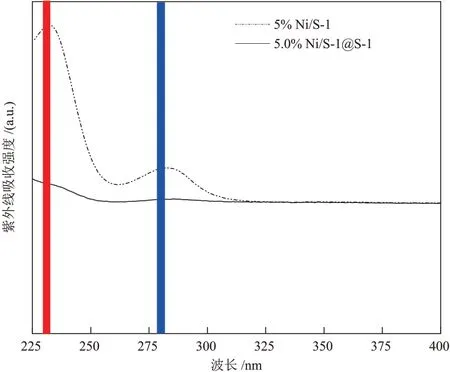
图5 不同催化剂催化间硝基甲苯加氢的紫外-可见光谱图Fig.5 Ultraviolet-visible spectra of different catalysts in catalytic hydrogenation of m-nitrotoluene
由表2可知,按nSiO2,S-1/nSiO2,硅溶胶= 1/1包覆后金属含量下降一半,说明内部S-1及NiO颗粒全部被包覆。
表2 不同催化剂的ICP元素分析结果Table 2 ICP elemental analysis results of different catalysts
2.4 催化剂还原特性分析
催化剂的还原性质对活性金属-金属氧化物的组成与结构具有重要影响。催化剂的H2-TPR 曲线如图6 所示(虚线为拟合结果,实线为测量结果),5% Ni/S-1 催化剂在426 ℃有一个明显的还原峰,对应表面NiO 物种的还原,而5.0% Ni/S-1@S-1 催化剂的还原峰位移至485 ℃,并且存在一个肩峰,对应在S-1 分子筛内部、与其具有较强相互作用的NiO 物种[16-17]。还原温度的显著提高表明S-1分子筛封装金属增强了NiO 与S-1 分子筛的相互作用,进而抑制其向Ni0还原,有利于保留更多具有Ni2+等具有高价态的氧化物。该类氧化物由于对H2及*CO 等中间物种的活化解离作用较弱,可被用于调控CO2加氢反应的加氢深度及产物的选择性[18]。
图6 不同催化剂的H2-TPR曲线Fig.6 H2-TPR curves of different catalysts
2.5 催化剂结构演变分析
2.5.1 预处理与反应过程中的结构演变分析
利用原位XRD 考察了5.0% Ni/S-1@S-1 与5% Ni/S-1催化剂在H2-预还原处理与CO2加氢反应过程中的催化剂结构演变,结果如图7 所示。由图7(a)可知,在H2-预还原处理过程中,随还原温度的升高,5.0% Ni/S-1@S-1 催化剂在2θ= 43.3° 处对应立方相NiO(No.02—1216)的(200)晶面的衍射峰逐渐减弱,而2θ= 44.7° 处对应立方相Ni0(No.01—1260)的(111)晶面的衍射峰逐渐增强。Ni0的衍射峰在320 ℃时出现,表明此时体相的NiO物种开始还原,而NiO的衍射峰在400 ℃还原30 min 后完全消失,将经过400 ℃ H2-预还原处理后的5.0% Ni/S-1@S-1催化剂记为R-5.0% Ni/S-1@S-1,进一步对R-5.0% Ni/S-1@S-1催化剂进行CO2加氢反应中的结构测试。由图7(b)可知,在CO2加氢反应过程中,包覆的S-1分子筛的拓扑结构依然保持稳定,且Ni0物种对应的衍射峰强度无明显变化,无其他类型Ni物种,如NiO、Ni3C等物质的特征峰[19]的出现,表明该情况下实际反应中的活性物种为Ni0。
图7 5.0% Ni/S-1@S-1催化剂在H2-预还原阶段的原位XRD谱图(t = 300~400 °C,p = 0.1 MPa)(a)和5.0% Ni/S-1@S-1催化剂在CO2加氢反应阶段的原位XRD谱图(t = 200~300 °C,p = 0.1 MPa,GHSV = 20000 mL/(g·h))(b)Fig.7 In situ XRD patterns of 5.0% Ni/S-1@S-1 catalysts during H2 reduction process (t = 300~400 °C, p = 0.1 MPa) (a)and in situ XRD patterns of 5.0% Ni/S-1@S-1 catalysts during CO2 hydrogenation process (t = 200~300 °C, p = 0.1 MPa,GHSV = 20000 mL/(g·h)) (b)
5% Ni/S-1催化剂在H2-预还原与CO2加氢反应中的原位XRD谱图如图8所示。还原温度升至260 ℃时,2θ= 44.7° 处出现立方相Ni0物种的衍射峰,随还原温度升高,其衍射峰逐渐增强。同时,在320 ℃时,2θ= 43.3° 处立方相NiO 的衍射峰完全消失,对应此时NiO 物种到Ni0物种的还原过程结束,Ni 物种全部以Ni0的形式存在,将400 ℃ H2-预还原处理后的5% Ni/S-1 催化剂记为R-5% Ni/S-1。相比于5.0% Ni/S-1@S-1催化剂,5% Ni/S-1中NiO向Ni0还原过程的起始(260 ℃)与结束温度(320 ℃)明显降低,与H2-TPR 曲线结果相一致,表明S-1 分子筛的封装增强了NiO 的稳定性。300 ºC 长时间反应后,依然仅观测到Ni0的衍射峰,表明该情况下其在反应过程中的活性相为Ni0。
图8 5% Ni/S-1催化剂在H2-预还原阶段(t = 260 ~ 400 °C,p =0.1 MPa)与CO2 加氢反应(t = 300 ℃,p = 0.1 MPa,GHSV = 20000 mL/(g·h))阶段的原位XRD谱图Fig.8 In situ XRD patterns of 5% Ni/S-1 catalysts during H2 reduction process (t = 260~400 °C, p = 0.1 MPa) and CO2 hydrogenation process (t = 300 ℃, p = 0.1 MPa,GHSV = 20000 mL/(g·h))
2.5.2 非预处理样品在反应过程中的结构演变分析
将不经过H2-预还原的5.0% Ni/S-1@S-1 与5% Ni/S-1 催化剂分别记为N-5.0% Ni/S-1@S-1 与N-5% Ni/S-1,直接进行CO2加氢反应测试,利用原位XRD考察其结构变化,结果如图9所示。由图9(a)可知,对于N-5.0% Ni/S-1@S-1 催化剂,在CO2加氢反应气氛(V(CO2)/V(H2)/V(N2) = 21/63/16 )中,随反应温度升高,NiO 的衍射峰基本保持不变,仅在380~400 ℃区间内观测到微弱的Ni0衍射峰,表明此时少量的NiO 物种被还原。在300 ℃,0.1 MPa 的CO2加氢反应中,N-5.0% Ni/S-1@S-1催化剂实际的活性相为NiO。由图9(b)可知,随反应温度升高,NiO逐渐向Ni0转化,在300 ℃,0.1 MPa的CO2加氢反应中,N-5% Ni/S-1催化剂实际的活性相为NiO与Ni0的混合物。通过XRD 结果进行半定量分析,此时Ni物种的组成为n(NiO)/n(Ni0) = 56.9%/43.1%。
图9 N-5.0% Ni/S-1@S-1 催化剂(a)和N-5% Ni/S-1 催化剂(b)在CO2加氢反应阶段的原位XRD 谱图(t = 200~400 ℃,p = 0.1 MPa,GHSV = 20000 mL/(g·h))Fig.9 In situ XRD patterns of N-5.0% Ni/S-1@S-1 catalysts (a)and N-5% Ni/S-1 catalysts (b) during CO2 hydrogenation reaction (t = 200~400 ℃, p = 0.1 MPa, GHSV =20000 mL/(g·h))
2.6 CO2加氢反应测试分析
对上述4 种催化剂,即400 ℃ H2-预还原后的R-5.0% Ni/S-1@S-1与R-5% Ni/S-1催化剂以及未经过H2-预还原的N-5.0% Ni/S-1@S-1 与N-5% Ni/S-1催化剂进行CO2加氢反应测试,结果如图10(a)所示。在相近的转化率下,活性相为Ni0的R-5.0% Ni/S-1@S-1与R-5% Ni/S-1催化剂主要产物为CH4,其选择性均超过98%。活性相为NiO 的N-5.0% Ni/S-1@S-1 催化剂产物则主要为CO,其选择性为96.4%。活性相为NiO 与Ni0混合物的N-5% Ni/S-1 催化剂中,CH4与CO的选择性分别为59.1%与36.1%,此外C2H6的选择性为4.8%,相对较高的C2H6选择性表明其形成所必需的碳链增长过程可能依赖于Ni0-NiO 界面结构。进一步对320 ℃与380 ℃下H2-预还原后的5.0% Ni/S-1@S-1催化剂(R(320)-5.0% Ni/S-1@S-1与R(380)-5.0% Ni/S-1@S-1)进行CO2加氢反应测试,分别测定不同预处理条件下的产物CH4与CO生成速率,将其与经由XRD谱图分析得到的n(NiO)/n(NiO + Ni0)进行线性关联,结果如图10(b)所示,CH4的生成速率随NiO含量的增大而线性下降,而CO的生成速率则随之线性升高。上述结果表明,通过调整S-1分子筛封装策略与预处理条件,可以实现NiO向Ni0还原的可控调节,进而控制CO2加氢深度,实现反应路径和产物(CO与CH4)选择性的可控调节。
图10 CO2加氢反应测试结果(t = 300 °C,p = 0.1 MPa,GHSV = 20000 mL/(g·h))(a)及n(NiO)/n(NiO + Ni0)与 CH4和CO生成速率的关系(b)Fig.10 Test results of CO2 hydrogenation reaction (t = 300 ℃, p = 0.1 MPa, GHSV = 20000 mL/(g·h)) (a) and relationships between n(NiO)/n(NiO + Ni0) and generated rates of CH4 and CO (b)
3 结论
本文采用浸渍-再晶化的策略,制备了S-1 分子筛封装Ni 基催化剂,通过调控Ni 物种的价态与结构组成以实现对CH4或CO 的选择性生成,得到如下结论。
(1)以SiO2纳米球和S-1 分子筛分别作为载体(Ni/SiO2@S-1和 Ni/S-1@S-1),SEM结果表明,相比于SiO2纳米球载体,S-1载体具有模板导向作用,可促进再结晶过程S-1 分子筛的生长,在较低TPAOH加入量(n(TPAOH)/n(SiO2) = 0.1)与较短晶化时间(24 h)下即可使封装S-1分子筛快速结晶,以实现对Ni 物种的良好封装。封装后的Ni/S-1@S-1 催化剂呈现规则的盒状结构,且S-1 包覆层的介孔结构提供了额外的比表面积。EDX 和大分子液相加氢实验结果证实了封装效果良好,Ni物种主要分布在包覆的S-1分子筛内部。
(2)H2-TPR 与原位XRD 结果表明,Ni/S-1@S-1催化剂增强的NiO 与S-1 载体间的相互作用,有利于抑制NiO向Ni0的还原,提高NiO在富氢环境(H2-预还原与CO2加氢反应)下的结构稳定性。
(3)通过调变催化剂的合成(封装或浸渍)与H2-预还原条件,可实现对NiO与Ni0物种组成比例的调控,n(NiO)/n(NiO + Ni0)与CO 的生成速率呈线性正相关,而与CH4的生成速率呈负相关,表明NiO 为CO 生成主要的活性中心,而 Ni0为CO2深度加氢形成CH4的活性中心。
上述工作为厘清Ni 基催化剂在CO2加氢反应中的本征活性中心,并调控活性结构组成,以实现加氢产物(CO 或CH4)的选择性生成提供了借鉴与依据。
