基于密集连接神经网络和长短期记忆网络的赖氨酸戊二酰化位点的预测
2023-10-12吕佩诺贾建华
吕佩诺,贾建华
(景德镇陶瓷大学 信息工程学院,江西 景德镇 333403)
0 引言
赖氨酸戊二酰化广泛存在于真核生物和原核生物中,戊二酰基团(如戊二酰辅酶A)通过酶的催化共价结合到赖氨酸残基上。赖氨酸戊二酰化主要发生在线粒体中[1],线粒体功能障碍可导致衰老和相关疾病,如癌症,代谢性、神经性疾病等[2]。赖氨酸戊二酰化将影响线粒体代谢及功能[3],目前已经证实了赖氨酸戊二酰化对阿尔兹海默症、帕金森病等神经退行性疾病的影响,以及对神经细胞的损伤[4]。2014年Tan等[5]首次通过生化实验在大肠杆菌和小鼠肝细胞中验证了赖氨酸戊二酰化位点,证明了脱戊二酰化酶(SIRT5)和营养物可以对赖氨酸戊二酰化进行调节。2016年,Xie等[6]利用实验方法从24条蛋白质中鉴定了41个赖氨酸戊二酰化位点。使用实验方法鉴定赖氨酸戊二酰化位点虽然准确但耗时耗力,需要寻找新的计算方法提高预测效率。
采用计算方法对赖氨酸戊二酰化位点的预测研究相对较少,这项工作具有挑战性。2018年Ju等[7]首次开发了赖氨酸戊二酰化位点的预测工具GlutPred,研究结合多种特征编码方法,采用偏置向量机模型处理数据的不平衡问题。同年,Xu等[8]采用氨基酸指数、K间隔氨基酸对组成、位置特异性氨基酸倾向、位置特异性倾向矩阵四种不同的特征编码方案,构建了iGlu-Lys预测器。iGlu-Lys性能高于GlutPred,但他们采用的数据集较小,且敏感性得分都较低。2019年Huang等[9]基于序列特征编码和最大依赖性分解(MDD)捕捉位置间的相互依赖,利用SVM算法构建了MDDGlutar分类器,该分类器所有性能比较均衡但准确率略低。Al-barakati等[10]利用序列编码方法和随机森林算法构建了RF-GlutarySite分类器,该模型使用了一个更大的数据集,但训练和测试时都是平衡数据集,不能很好地反映真实情况。2020年,Dou等[11]开发了基于AdaBoost算法和三种特征编码方法的iGlu-AdaBoost分类器,采用混合采样法对训练数据集进行处理,该测试数据集是非平衡的。2022年Liu等[12]提出了深度学习预测算法,该算法基于词嵌入和深度神经网络框架搭建而成,评估了不同单词嵌入和不同深度学习模型的预测性能。2022年Qiao等[13]针对不平衡数据采用SMOTE和Tomek Links筛选重组数据,应用六种特征编码方法并基于XGBoost算法建模构建了DEXGB_Glu预测器。同年Indriani等[14]将传统的基于序列的特征提取方法与预训练的transformer的特征提取模型相结合,筛选出了性能最好的分类器ProtTrans-Glutar。尽管目前关于赖氨酸戊二酰化位点已经开发了一些预测工具,但和其他位点相比,赖氨酸戊二酰化的预测模型相对较少,且性能不够理想,准确性和真实性还有很大的提升空间。
越来越多的研究表明,深度学习方法可以成功运用在蛋白质位点预测上。过去应用在预测赖氨酸戊二酰化位点的深度学习方法主要有卷积神经网络和长短期记忆网络(Long Short Term Memory,LSTM[15])等,类似的方式,密集连接神经网络(Densely connected convolutional networks,DenseNets[16])开始用于挖掘其他氨基酸序列信息,并且实现了比传统模型更好的性能。本研究中引入密集连接神经网络和长短期记忆网络,提出了一种新的预测方法来鉴定蛋白质中的赖氨酸戊二酰化位点。实验表明,对序列做简单编码后再利用深度学习模型提取特征信息,不仅能提高数据处理的效率,还能提高预测的性能。
1 材料与方法
1.1 基准数据集
在这项研究中采用了Al-barakati等人[10]构建的非平衡数据集。该数据集来源于蛋白质赖氨酸修饰数据库(PLMD)、SWISS-PROT数据库以及国家生物技术中心(NCBI),包括四种不同物种(小家鼠、结核分枝杆菌、大肠杆菌和HeLa细胞),共有234个蛋白质的749个位点。使用CD-Hit[17]去除了同一性大于40%的同源性序列,滑动窗口提取序列片段,得到长度为23的肽序列。最后保留了400个阳性位点和1703个阴性位点作为训练集,44个阳性位点和203个阴性位点作为独立测试集。如表1所示:

表1 基准数据集
1.2 one-hot编码
在这项工作中使用one-hot编码[18]方式对蛋白质序列进行编码。这是一种离散型表示,一条序列中氨基酸对应的指数为1,其他位置为0。如丙氨酸的编码为10…0,半胱氨酸的编码为01…0。总共有20个氨基酸个数,加上未知项X,one-hot编码的长度为21。对于长度为L的序列片段,最终得到L*21维向量。本研究的数据序列长度L=23,得到一个23*21维度的矩阵。
1.3 模型结构
本研究建立了一个深度学习模型去预测赖氨酸戊二酰化位点。在这个模型中,通过密集连接的卷积块和LSTM层进行特征提取后得到相关矩阵,输入到两个层数为300层的全连接层中,最后利用softmax层进行分类,从而有效地预测赖氨酸戊二酰化位点。Glu-DClstm整体模型如图1所示:
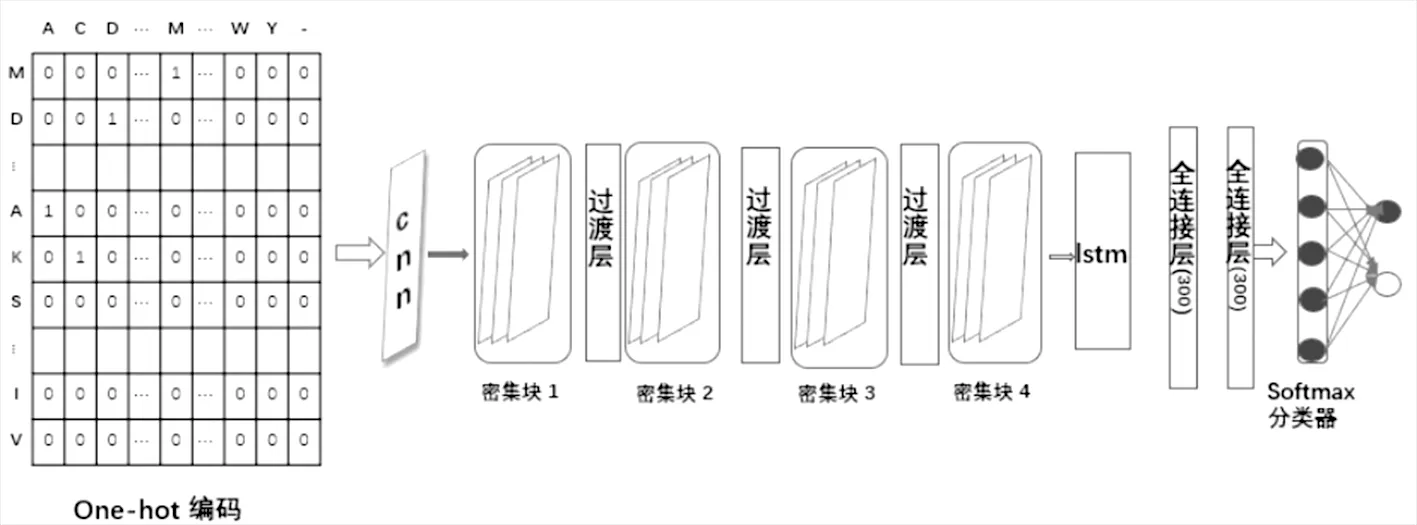
图1 Glu-DClstm模型结构
1.3.1密集连接神经网络
密集连接神经网络的密集连接机制,在一定程度上减轻了梯度消失,加强了特征的传递,在参数与计算量更小的情况下取得更优良的性能,优于传统的CNN和ResNet[18]。
在应用密集卷积块之前,首先输入one-hot编码矩阵,通过一维卷积层生成序列信息的低级特征信息图。如公式(1)所示:
h0=a(E×W+b)
(1)
其中,E是one-hot编码,one-hot编码的长度为21。W是权重矩阵,大小为21×S×D,S是卷积核的大小,D是卷积核的数量。S=4,D=96,b是偏置项,h0是一维卷积层的输出,大小为L×D。a是ELU激活函数,实现非线性变换。
特征编码经过卷积层后的输出向量是密集卷积块的输入向量,密集卷积块对上一层的信息进行一系列的卷积运算后获得高级特征表示图。如公式(2)所示:
hk=a([h0;h1;…;h(k-1)]×W′+b′)
(2)
其中,hk-1表示密集卷积块中第(k-1)个卷积生成的特征向量,W′∈RD′×S×D″是权重矩阵,D′由K决定,D″是每一层卷积核的数量,这里设置为32,b′是偏差,[h0;h1;…;hk-1]表示将密集卷积块的输出h0;h1;…;hk-1沿特征维度串联。
然后,在两个密集卷积块之间使用一个过渡层对密集卷积块输出的高级特征图进行卷积和池化操作。如公式(3)所示:
hk=a([h0;h1;…;h(k-1)]*W′+b″)
(3)
W′∈R(D′+D″)×S′×(D′+D″)是权重矩阵,S′是卷积核的大小,设置为1,b″ 是偏置项,最后对hk采用平均池化操作降维。
将多个密集卷积块和过渡层串联起来即构建出密集连接神经网络,本研究中,设置了4个密集卷积块,最终可以提取蛋白质序列的高级特征。
1.3.2长短期记忆网络
长短期记忆网络(LSTM)是一种特殊的递归神经网络(recurrent neural network,RNN),相较于传统的RNN,LSTM解决了其梯度爆炸和梯度消失问题[15]。LSTM主要由三个门(遗忘门、输入门、输出门)、细胞态(cell state)、记忆体和候选态构成。其中细胞态是LSTM的核心,也是与RNN的最大区别之处。细胞态负责数据的长期记忆,数据将沿着细胞态流动。三个门能控制细胞态中信息的增加或移除,结构如图2所示。

图2 LSTM结构
其中,t时刻的隐藏状态为ht,细胞态为Ct,输入特征Xt。t-1时刻的隐藏状态为ht-1,细胞态为Ct-1。σ是sigmoid激活函数,tanh为tanh激活函数。ft代表遗忘门,it代表输入门,ot代表输出门。
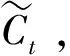
1.4 评估指标
本研究中采用十折交叉验证和独立测试集来评估模型的性能。十折交叉验证是将训练集平均分成十个子集,其中九个用于训练,一个用于测试,重复十次直到每个子集都作为一次测试集,十次训练的平均结果为训练集结果。同样,独立测试集的结果也用来评估构建的模型。本研究中计算了四个统计指标:敏感性(Sn),特异性(Sp),准确性(Acc),马修斯相关系数(MCC)。这些指标的公式如下:
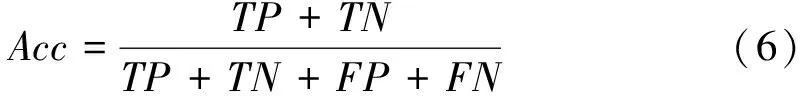
其中TP、TN、FP、FN分别表示真阳性(正确预测赖氨酸戊二酰化位点的数量)、真阴性(正确预测非赖氨酸戊二酰化位点的数量)、假阳性(错误预测赖氨酸戊二酰化位点的数量)和假阴性(错误预测非赖氨酸戊二酰化位点的数量)。Sn是用来衡量阳性准确率即识别赖氨酸戊二酰化位点的准确性的指标,Sp是用来衡量阴性准确率即识别非赖氨酸戊二酰化位点的准确性的指标,Acc代表分类正确的样本占总样本个数的比例[20]。MCC可以合理评估非平衡数据集下的二元分类模型的效果,MCC越高,证明该分类模型对非平衡数据的预测效果越好[20]。另外,还测量了受试者特征(Receiver Operating Characteristic,ROC)曲线和曲线下的面积(Area under ROC,AUC)。AUC值越接近1,表示分类器精度越高[21]。
2 结果分析
2.1 十折交叉验证
在本研究中,构建了Glu-DClstm模型预测戊二酰化位点,选择了目前最新的数据集之一。Glu-DClstm模型采用one-hot编码,结合密集连接神经网络和长短期记忆网络模型挖掘序列信息。本研究采用十折交叉验证和独立测试集评估了模型的Sn、Sp、Acc、MCC和AUC的性能。
Al-barakati等人构建的RF-GlutarySite模型将数据集提前处理为平衡数据集,而本研究构建的模型是在非平衡数据集下训练和预测的。因此为了保证比较的准确性,本研究同采用非平衡数据的ProTrans-Glutar模型进行了比较。
另外为了进一步验证密集连接神经网络和长短期记忆网络组合模型的优势,本研究尝试去掉LSTM模型,单独对DenseNets模型进行验证。十折交叉验证的结果如表2所示,DenseNets和LSTM的组合模型效果更加均衡且优良,Sn,Sp,Acc,MCC分别为0.6750、0.6735、0.6738、0.2842,高于目前最新的预测器ProTrans-Glutar。Glu-DClstm具有稳健性,AUC为0.744,ROC曲线如图3所示。
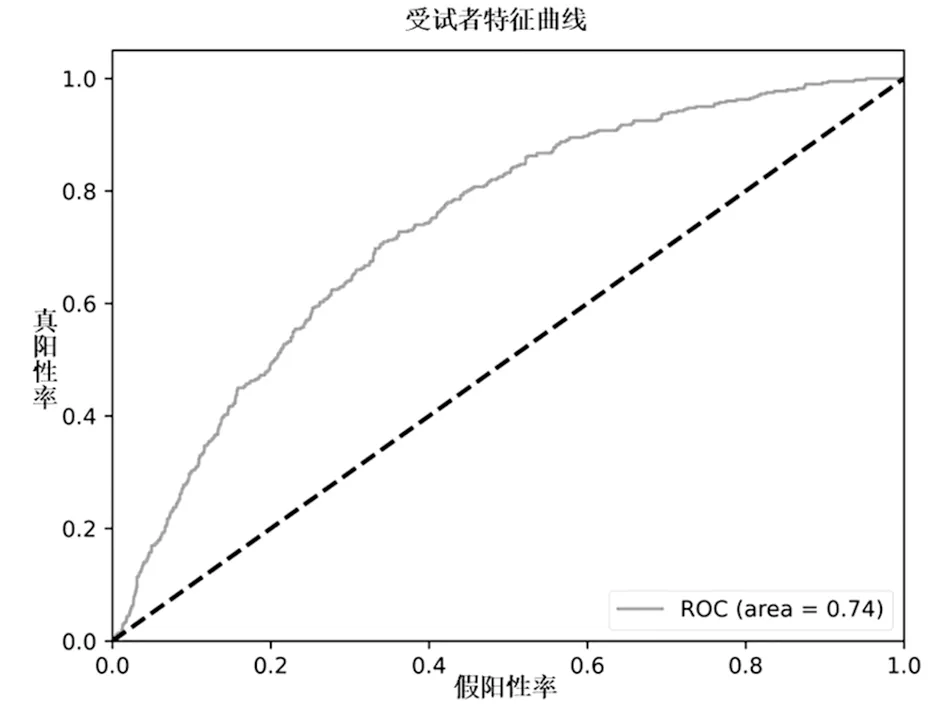
图3 训练集ROC曲线
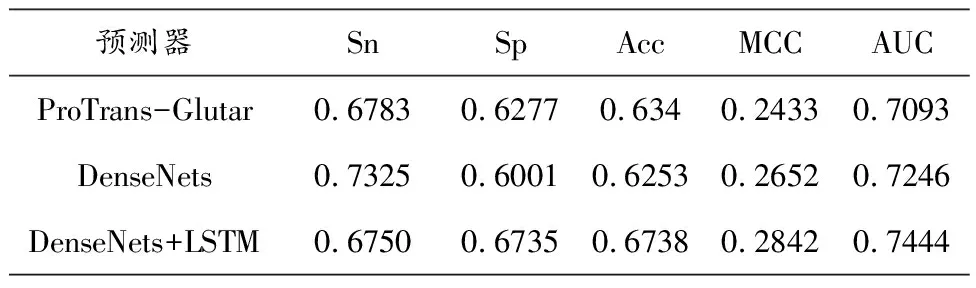
表2 十折交叉验证结果
2.2 独立测试集与现有模型的比较
为了验证构建的模型具有良好的泛化能力且优于其他预测模型,本研究将独立测试集的结果与其他的预测器进行了比较。为保证模型不会因为训练集不同而造成结果偏差,本研究采用了相同的独立测试集分别对GluPred、iGlu-Lys、MDDGlutar、iGlu-AdaBoost、ProtTrans-Glutar进行对比。结果如表3所示,GluPred和iGlu-Lys虽然Acc较高,但其Sn只达到了随机预测水平,模型实际意义不大,不具有可参考性。Glu-DClstm的Sn和Sp的值更均衡,相比Sn低Sp高的模型更具有实际意义。Glu-DClstm与MDDGlutar、iGlu-AdaBoost、ProtTrans-Glutar相比Sp至少提高了3%,Acc至少提高了4%,MCC至少提高了1%,AUC至少提高了9%。ROC曲线如图4所示。
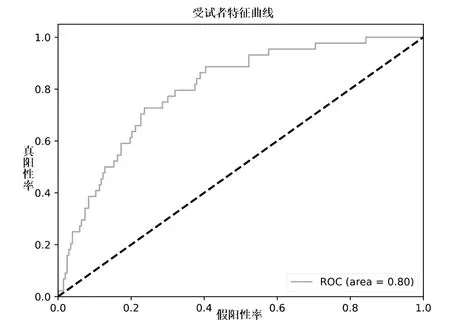
图4 独立测试集ROC曲线
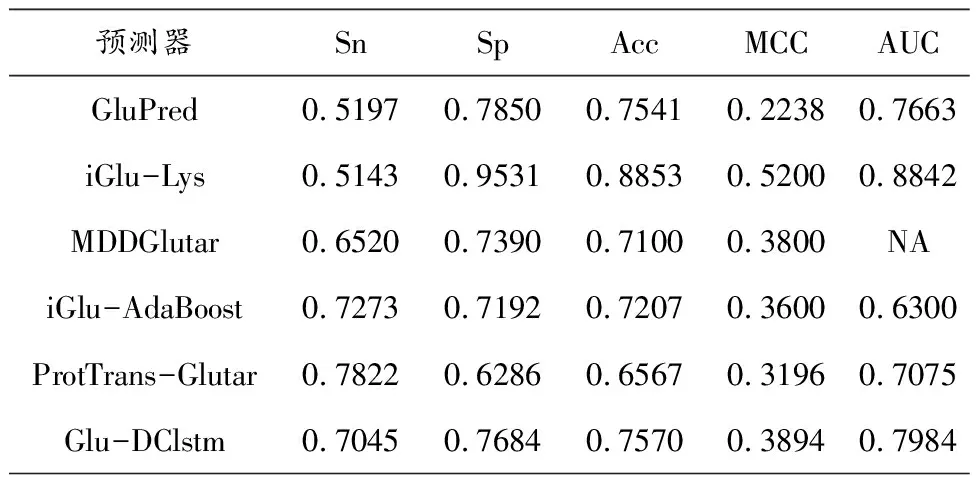
表3 独立测试集对比结果
如表3所示,Glu-DClstm的预测结果:Sn=0.7045、Sp=0.7684、Acc=0.7570、MCC=0.3894、AUC=0.7984,与最新的预测器ProtTrans-Glutar相比,Glu-DClstm 的Sp提高了14%,ACC提高了10%,MCC提高了7%,AUC提高了9%。尽管Sn略有降低,但总的来说Glu-DClstm模型对赖氨酸戊二酰化位点预测更平衡,MCC和AUC作为整体模型性能的评估指标,在对非平衡数据集的预测时,研究者更关注MCC和AUC的值是否有所提高。其他预测器的敏感性和特异性差异较大,在实际运用中容易造成较大的误差。Glu-DClstm模型解决了这个问题,给出了一个比较平衡的结果,优于现有的预测模型。本研究的数据集样本正负比例约为1∶4.5,数据较不平衡。Glu-DClstm在非平衡数据集的处理上采用类权重的方法,避免了直接采样带来的信息损失,对赖氨酸戊二酰化位点预测能提供较大帮助,更具有实用价值。
3 讨论
本研究首先对蛋白质序列进行one-hot编码,利用密集连接神经网络和长短期记忆网络模型充分挖掘序列信息,建立了一种新的预测赖氨酸戊二酰化位点的模型。同时独立测试集的评估表明,该模型对正负样本数据不平衡问题的处理是有效的。正负样本的预测结果比较均衡,与其他预测器相比该模型表现更加优良,具有良好的泛化能力。
另外,一个公开友好的在线预测网站将大大提高研究效率,因此在未来工作中,将致力于建立一个公开稳定的Web在线服务器,为广大研究者提供便利。
