第35届中国化学奥林匹克(初赛)试题解析
——以有机化学部分第9、10题为例
2023-10-07杨明岸李俊吴敏锶张喜庭
杨明岸,李俊,吴敏锶,张喜庭
1 广东第二师范学院化学系,广州510303
2 华南师范大学化学学院,广州 510006
3 广州大学化学化工学院,广州510006
第35届中国化学奥林匹克(初赛)在全国28个省、自治区、直辖市举行,约7万名中学生参与了本次竞赛[1]。作为全国性的化学赛事,竞赛鼓励青少年接触化学发展的前沿,激发学生钻研化学的兴趣,探索化学家的思想渊源与工作方法,为国家选拔化学理论功底扎实、能与化学家在研究思路上产生“共振”的科学型人才创造了机遇与条件[2]。总体来看,试题的综合难度与往届持平。相较于第34届试题,在知识板块的分布上,分值比重明显增加的是无机化学(含结构化学,如分子结构、晶体结构);明显减小的是高分子化学、配位化学与分析化学;基本持平的是元素化合物、有机化学与物理化学。初赛的基本内容仍集中在无机化学(含元素化合物)和有机化学这两个部分,分数约占全卷的60%[3]。有机化学模块在试题的设置上稳中求进,侧重于考查学生对基础知识的理解与运用能力,部分内容选材于近两年有机化学领域较新并承载重要意义的研究成果。此外,试题强化了空间力、推理力与信息分析能力这三个维度的要求。通过对第35届化学初赛第9、10题的分析(见表1),帮助处于启蒙阶段的竞赛选手熟悉有机化学模块的试题结构与考查风格,为从事化学竞赛培训的教师提供参考。第35届中国化学奥林匹克(初赛)有机化学部分第9、10题原题已放入补充材料,读者可从参考文献[1]或本文的补充材料快速获得竞赛题目。

表1 第35届化学奥林匹克(初赛)第9、10题考情细目表
1 第9题解析
1.1 考题解析
[9-1] 本题考察吡啶类衍生物与酸酐、三级膦之间的反应。反应的转化过程与第34届初赛第10题的Pummerer重排较接近[2]。二芳基三氟甲基膦是一种亲核试剂。结合产物结构看,它不与Tf2O (三氟甲磺酸酐)反应,而是对吡啶进行三氟甲基化。Tf2O起到活化吡啶环的作用,环上的N原子为亲核位点,Tf2O的S原子为亲电位点,发生加成反应生成中间体M1。在O-的参与下,通过给电子对的转移,TfO-作为离去基团发生消除反应。二芳基三氟甲基膦亲核进攻中间体M2的C(4)进行1,4-加成反应,生成具有醌式结构的季鏻盐,为吡啶结构的三氟甲基化提供了可能性。中间体M3的C(4)的质子被大位阻碱DBU (1,8-二氮杂双环[5.4.0]-7-十一碳烯)[4]攫取转化为具有亲核能力的反应位点,在给电子对的作用下,N-S键断裂使Tf-基团离去。由于是强酸性水解的条件,中间体M4吡啶环上的N在质子化的同时,磷原子结合水分子中的羟基。经分子内重排,离去二芳基羟基膦,最后得到三氟甲基化产物。反应的历程如图1所示,A和B的结构简式见图2。

图1 吡啶C4位三氟甲基化的反应机理

图2 化合物A和B的结构简式
[9-2] 本题考察Claisen重排反应与Alder-ene反应在成环中的应用。反应条件是330 °C的高温,无外加试剂,可联想到周环反应。底物是一个烯丙基乙烯基醚类衍生物结构,是典型的[3,3]-σ重排的骨架体系,故由反应物转化到中间体M5发生的是Claisen重排反应(图3标注的两个M5是同一种中间体,即题目要求书写的A。为表示清楚反应历程,凸显出不同位置的H原子)。反应经历六元环过渡态的分子内重排,烯氧基在五元环纸面朝下,所以协同反应产生的乙醛基仍在五元环纸面朝下。在完成[3,3]-σ重排后,发生周环反应。周环反应过程涉及的位点如图3所示(红色突出)。从M5到pdt-1,存在的转化有:① H原子由C(5)转移到C(9);②π键断裂:C(1)-C(2),C(8)-C(9);③ 新键形成:σ键C(2)-C(8),π键C(1)-C(5);从M5到pdt-2,存在的转化有:① H原子由C(10)转移到C(1);②π键断裂:C(1)-C(2),C(8)-C(9);③ 新键形成:σ键C(2)-C(8),π键C(9)-C(10)。根据上述信息发现这与一般的周环反应类型如电环化、σ迁移的特征不符,与环加成(如D-A反应)具有相当的共性。本题中,电子从烯体(含烯丙基氢的富电子基团)的HOMO流向亲烯体(含π键的缺电子基团)的LUMO。过程涉及H原子与烯体双键的迁移,在亲烯体不饱和键的两端构建两个新的σ键,同时双键移动至原烯丙基位置,这一转化路径被称为Alder-ene反应(烯反应),属于六电子的周环过程。C(1)-C(2),C(8)-C(9)这两个双键的α-C上均有一个H原子,进行烯反应时两个双键都具备提供烯丙基氢的潜在性,故存在两种产物:若C(2)-C(1)-C(5)体系作为烯体,C(8)-C(9)作为亲烯体,生成物是pdt-1;若C(8)-C(9)-C(10)体系作为烯体,C(1)-C(2)作为亲烯体,生成物是pdt-2。本题中烯反应的环状过渡态为稳定的船式构象TS-1和TS-2[5],分别对应题目的过渡态B和C;M5对应题目的中间体A,转化过程见图3。

图3 烯丙基乙烯基醚类衍生物在高温条件下进行Claisen重排与Alder-ene反应的机理
[9-3] 本题涉及亲核取代反应、核磁共振氢谱和芳香性等重要知识。一般来说,进攻的亲核试剂的亲核能力越强,反应经过SN2机理过渡态所需的活化能就越低,SN2反应趋向越大。AgNO3是一种弱的亲核试剂,故倾向于进行SN1反应。反应物中的Cl原子被Ag+以静电作用诱导离去,生成的碳正离子被-ONO亲核进攻得到了产物硝酸酯[6,7]。而MeLi是一种强的亲核试剂,倾向于进行SN2反应。
根据题意“分子式为C9H12的产物”,结合反应物的分子式C8H9Cl后可推断出氯原子被换作甲基。“产物的1H-NMR谱表明,在化学位移0.89 ppm处有六个氢”可分析出:① 由于六个氢原子是化学等价的,产物中很可能存在两个化学环境一致的甲基;② 质子吸收峰处于0.89 ppm的信号位置是光谱中屏蔽效应很强(可通俗理解成质子对外磁场的感受少)的高场区域。从数值上看,甲基所连基团的给电子能力应比较强。反应物中已有一个甲基,可推断出第二个甲基是由试剂MeLi亲核进攻得到的。甲基负离子(Me-)的加成将得到碳负离子中间体,可将甲基负离子加成到各种位置,对得到的各种碳负离子中间体进行比较。如图4所示:由Hückel规则得知,环戊二烯负离子具有芳香性,稳定性好,证明了Me-进攻C(6)的反应倾向于发生。接着环戊二烯负离子进攻C(8)-Cl键使其异裂,Cl-离去,发生分子内关环。产物中两个甲基均连在环丙基上,印证了上述的推断②。
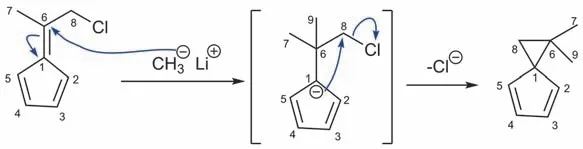
图4 环戊二烯衍生物和甲基锂发生SN2反应的机理
1.2 评注与拓展
[9-1] 本题可从分子式与价态的角度思考:将两个底物的分子式相加并与A的分子式比较。A相比于底物多了1个C,1个S,3个F,3个O原子;少了1个H原子。多出的原子部分可恰好组成TfO-,作为A的阴离子。减少的氢质子很可能被DBU捕获,形成DBUH+作为B的阳离子(DBUH+的分子式不符合A中阳离子的要求)。由“吡啶的C4位三氟甲基化”可知,二芳基三氟甲基膦倾向于亲核进攻C(4),在吡啶环上引入鏻鎓离子。此时P(III)变为了P(V),发生氧化反应。相应地,Tf2O就被还原成Tf-,可作为B的阴离子。经检验,这一推断满足得失电子守恒。A的阳离子骨架不难推断,可看成是鏻鎓离子取代吡啶C4位的H原子。
题目选材于2021年4月发表于Nature的一项研究成果[8]。科罗拉多州立大学的McNally课题组通过磷介导的sp2-sp3配体偶联反应,无需预官能团化和导向基团,便可通过“一锅法”直接将吡啶结构中的C-H键氟烷基化。该反应的选择性高,在复杂分子的后期功能化上展现出诱人的应用前景。
[9-2] Claisen重排反应的机理[9]如图5所示(该图不表达任何特定的前线轨道,只体现构象中新的成键与相应轨道的联系),这种[3,3]-σ迁移重排一般经过类椅式过渡态[10]910。

图5 Claisen重排反应的机理
化学家Alder在1943年首次提出Alder-ene反应。他通过高温加热丙烯和马来酸酐两种底物,合成了烯丙基琥珀酸酐,该过程可视为丙烯(含烯丙基氢)和马来酸酐发生了间接取代加成反应[11]。Alderene反应在大多数教科书中被归为环加成反应,在有些教科书中被独立划分为第四类周环反应[10]894-895,第30届决赛理论试题的[8-2]就涉及了这种反应[12]。看待Alder-ene反应最简单的方式,是将它转化为一类熟悉的环加成反应:D-A反应,其中双烯的一个π键被一个C-H键替代,产物形成一个新的C-C键与一个跨空间转移的H原子,反应机理见图6[13,14]。与Diels-Alder反应不同,Alder-ene反应中C-Hσ键的断裂要求更高的活化能,反应条件较为苛刻。但因其广泛的底物适用性[15],是一种用于高效构建C-C或C-X键(X:杂原子)的策略,在一些天然产物与复杂分子的合成中承担着重要的功能。

图6 Alder-ene反应的机理
[9-3] 核磁共振(NMR)波谱是化学家们进行结构解析与研究相互作用最常用的重要工具,是少数能用于无损检测三种物态的技术之一,灵敏度高(能分析质量不足1 mg样品的结构)。在解析物质的结构时,通常需要借助多个核磁实验从不同角度来展示分子中原子核及其核外电子的磁特性,据此信息推导出相应的分子结构。可以说,几乎所有的有机或生物分子以及许多无机分子的结构解析都源于NMR波谱技术[16]。自问世半个多世纪以来,NMR历经多次技术革新,这一领域的重大进展日新月异。本届初赛试题没有对NMR波谱单独命题,而将其作为辅助信息融入题干,彰显现代仪器定性分析的表征手段。“化学理论与实验结合”这一思想的渗透,是试题的特色之处。
2 第10题解析A
2.1 考题解析
[10-1] 本题考察酰胺共振式的书写。共振式之间的差别体现在电子的排布。书写分子的共振式时要注意:① 共振式要符合Lewis结构式;② 代表同一分子的共振式具有相同的原子排列顺序且具有相等的未成对电子数[17]。普通酰胺A的共振式可仿照烯醇互变异构书写,如图7采用共平面的画法。

图7 酰胺的共振式
[10-2-1] 本题考察端基效应与基团的电负性、离去能力之间的关系。基团Y的孤对电子偏移至N-Y之间,从而促使基团Z离去。Z基团的吸电子能力越强,越容易离去,使酰胺N原子的正电性增强,亲核试剂更容易与其发生取代反应,端基效应更强。本题中,基团的吸电子能力可通过电负性衡量,电负性有:F > O > N,即基团的端基效应有:F-> OR-> NR-2。故三种杂原子中,F原子取代酰胺具有最强的端基效应。
[10-2-2] 本题考察不同类型电子效应的竞争作用以及离子的稳定性。当酰胺转化为磺酰胺时,N原子左侧的酰基就变成了磺酰基。强的端基效应,意味着Z的离去能力更好。与N原子直接相连的酰基换成了吸电子能力更强的磺酰基后,就会与基团Z之间发生更强的吸电子竞争作用,从而削弱了端基效应。
[10-3] 本题考察加成-消除反应、自由基反应与氮宾的基本知识。tBuCOO-的离去能力强于BnO-,可确定tBuCOO-相当于[10-2]酰胺B中的离去基团Z。在-OBn孤对电子的参与下,通过给电子对的转移,tBuCOO-基团离去,发生消除反应。底物二级胺的N原子提供孤对电子,亲核进攻酰胺N原子,N=O间的一对π电子向O原子偏移,得到氮正离子中间体M6。氢质子被攫取后,受中间体M7内连着亚甲基的N的孤对电子影响,BnO-基团离去,产生中间体M8。接下来BnO-重新进攻带正电的酰基碳,经加成-消除反应后,得到二氮烯中间体M9。C-N键断裂的同时,释放出N2物种,生成了两个相同的含Br自由基,两者迅速结合形成产物。反应的机理如图8所示,转化过程中必定经过的两个关键中间体分别为M7和M9。
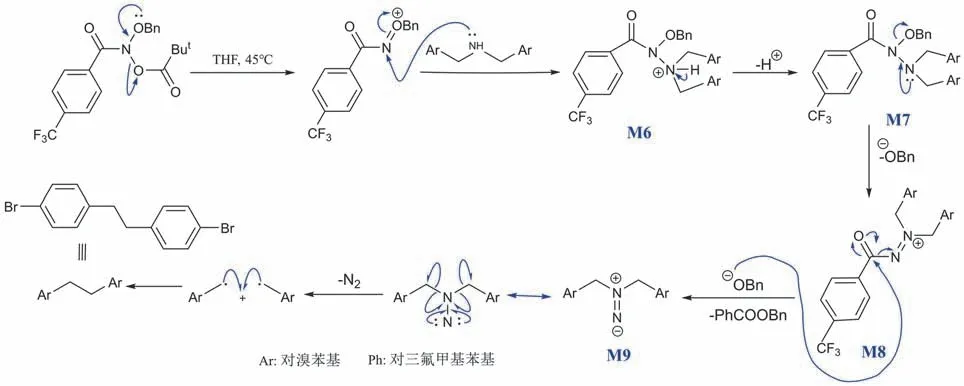
图8 具有端基效应的酰胺对二级胺骨架进行编辑以构建C-C键的反应机理
2.2 评注与拓展
[10-2-1] 用分子轨道理论(MO)解释本题,关键点是n(Y)和σ*(N-Z)两个轨道间的相互作用[18]。从N、O到F,随着Z原子的电负性依次增加,σ*(N-Z)轨道的能量逐渐降低,更加接近n(Y)轨道的能量;此外σ*(N-Z)轨道中N的成分逐渐增加,n(Y)与σ*(N-Z)轨道重叠的程度越大,体现了端基效应的调控作用,促进了化学反应的发生。端基效应(异头效应)属于静态立体电子效应。关于立体电子效应,可简要概括为:分子内的成键或非键电子在空间的特定方向产生轨道相互作用,进而引起分子的结构(含键长和键角)、能量与反应活性的变化[19]。这种效应可用于构象分析、立体化学以及反应过渡态的研究,是科学家在设计有机合成路线时不容忽视的因素之一[20]。
[10-2-2] 本题可从离子稳定性的角度考虑:基团Z离去后,N原子带正电,左侧酰基的吸电子作用不利于阳离子的稳定存在。当吸电子能力更强的磺酰基取代酰基后,形成的阳离子稳定性更差,基团Z就更难以离去,端基效应减弱。用MO来看:强吸电子基团可使N-Z发生极性反转,由此改变N对σ*(N-Z)轨道的贡献度。转化为磺酰胺后,N原子带δ-,Z原子带δ+,σ*(N-Z)轨道中N的成分下降,n(Y)与σ*(N-Z)轨道的重叠性变小,不利于化学反应的进行。
[10-3] 题目选自2021年5月发表于Nature的一项研究成果[21]。C-H键官能化反应虽然发展迅速,但能直接用于潜在分子骨架修饰的案例不多,骨架转化仍是一类尚未完全开发但发展潜力巨大的新型化学反应。从简化逆合成步骤的角度出发,实现对单原子“插入”与“删除”的操纵相当诱人。芝加哥大学化学系的Levin课题组发展了一种新型的分子编辑反应:直接“删除”仲胺中的氮原子,借助异二氮烯中间体释放出N2,实现分子内偶联以构建C-C键。反应的关键在于某种异头酰胺试剂可促进脂肪族仲胺的分子间活化。分子编辑技术有助于开发真正意义上的“无痕”化学反应——对合成有帮助的分子特征不残留在产物中,为有机分子结构的优化提供了新的思路[22]。
3 第10题解析B
3.1 考题解析
[10-4] 本题考察Dyotropic重排反应在复杂分子合成中的作用。
[10-4-1] 该反应的驱动力是通过扩环释放四元环的角张力。一个内酯环在扩张的同时,伴随着另一环的缩合。C(4)与C(5)分别连接的酰氧基同时处于反式共平面,具备协同迁移的倾向。理论计算表明这种Dyotropic重排由TMS+基团引发,类似于双重SN2反应[23-25],见图9。

图9 双内酯通过双重重排反应合成螺内酯
[10-4-2] 反应的驱动力仍然是四元环张力的释放,故C(6)-C(7)右侧迁移的是酰氧基。至于左侧迁移的是σ(C-H)还是σ(C-C),可从以下角度思考:① 若迁移σ(C-H),形成的是六元环。若迁移σ(C-C),则形成七元环,七元环结构的稳定性低于六元环;② 迁移σ(C-C)要求C(6)-C(7)左侧的烷基与右侧的酰氧基同时处于反式共平面,而这种构象的基团斥力作用大,稳定性差,因此在整个构象体系中所占的比例低。相应地,氢与酰氧基在纸面上处于反式的构象稳定性好;③分析过渡态TS-3和TS-4的构象[26],见图10。从空间效应的角度,TS-3的位阻相对较小,反应沿路径a进行的可能性大。但DFT(密度泛函理论)计算表明:TS-3与TS-4两种构象的能量较为接近。由于反应物具有构象灵活性,故空间效应不是唯一的决定因素。再者,σ(C-H)键轨道的能量通常比σ(C-C)键轨道能量高,且TS-4中σ(C-H)与σ*(C-O)之间轨道重叠的匹配性更好。综合上述因素,σ(C-H)键更倾向于迁移。反应的历程如图11所示。

图10 C-C键与C-H键迁移的化学选择性
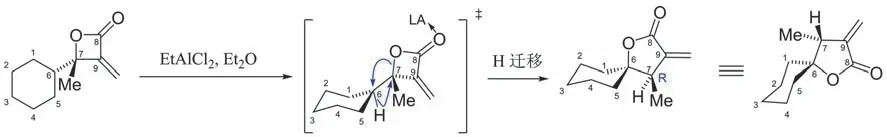
图11 α-亚甲基-β-内酯的双重重排反应
[10-4-3] 锂代二氢呋喃与二甲基铜锂(Gilman试剂)之间转金属化,在呋喃环上引入含Cu基团,构建了C-Cu键的分子骨架[27,28]。高阶铜酸盐中间体M10经过环状过渡态,Cu上的甲基和烷氧基团相互迁移形成中间体M11,水解后得到产物。重排的驱动力可能为“强碱制备弱碱”,反应历程见图12。

图12 有机铜试剂参与的双重重排反应
[10-4-4] 含Pd基团与苯基之间相互迁移,生成的中间体在金属Pd(II)的作用下还原消除直接得到非对映选择性产物,重新构建了C-F键。重排的驱动力可能为:① 空间相互作用的弱化;② 生成了热力学更稳定的α-羰基Pd(IV)物种[29]。反应的历程见图13。

图13 金属Pd基团参与的双重重排反应
3.2 评注与拓展
[10-4-2] 选自2020年12月发表在AngewandteChemieInternationalEdition的一项研究成果[26]。清华大学的唐叶峰课题组借助环张力驱动的Dyotropic重排反应,构建了一种合成α-亚甲基-g-丁内酯的通用方法。反应中迁移基团的迁移顺序具有一定的规律性,可实现产物的取代类型和立体化学的精准调控。研究指出该反应具有广泛的底物适用性,已被成功应用于一些天然产物及其核心骨架、生物探针分子等的合成,在某种程度上解决了潜在的副反应易发生、化学选择性难受控的问题,凸显了方法的实用性。
[10-4-3]与[10-4-4]选自NatureChemistry在2021年7月刊出的一篇文章[29]。洛桑联邦理工学院的祝介平团队成功实现了在温和的条件下邻位C-C键和C-Pd(IV)键的Dyotropic重排,并构建了一系列含有三个立体中心的氟化环戊烷,反应具有高度的立体专一性。这种重排过程为常规手段难以实现的C-C键活化创造了新的开端,且有望作为Pd催化反应的一种工具。
4 结语
第35届中国化学奥林匹克(初赛)有机化学部分的试题难度与往届相比变化不大,但立体化学的复杂性提升,涵盖的知识面更广,对选手的综合能力与高阶思维提出了进一步的要求。学生在日常训练中可参考国内外的著名期刊或高等有机化学书籍,加强反应机理的理解与推导。此外,鼓励学生接触化学发展的前沿,拓宽自身的视野,培养分析、运用信息解决综合问题的能力与创新型思维。
