SFOD-LPME-HPLC法测定白酒中甘草酸、甘草次酸
2023-09-28黄何何胡朝阳蔡小明
黄 媛,洪 丽,黄何何,潘 城,胡朝阳,蔡小明*
(福建省产品质量检验研究院 国家加工食品质量监督检验中心,福建 福州 350001)
白酒被誉为世界六大蒸馏酒之一[1],源自我国传统的酿造工艺,具有醇香甜美的独特口感。而现代生产工艺追求效益,大多发酵时间短,在白酒制作过程中产生多元醇或酮等天然甜味物质一般都达不到理想的效果,部分不良商家为了以次充好,在白酒中添加甜味剂来掩盖苦味[2]。我国标准GB 2760—2014《食品添加剂使用标准》中明确规定,白酒中不得添加任何甜味剂[3]。关于白酒中检出甜蜜素、糖精钠、安赛蜜等常见甜味剂的报道屡见不鲜[4-6],随着近几年监管的加强,对于白酒中违规添加人工甜味剂的现象已经越来越受到重视,为了逃避监管,商家极可能选择新型的不在监测范围内的甜味剂进行使用[7]。因此对于能够应用于白酒中的天然甜味剂也应当引起相关部门的关注与重视。
甘草酸(glycyrrhizin,GA)、甘草次酸(glycyrrhetinic acid,GTA)属于三萜类化合物,甜度约为蔗糖的80~300倍[8]。因其高甜、低热、安全的特点,在食品工业中作为甜味剂得到了广泛的应用[9-10]。鉴于其优越的甜度,在食品中添加少量即可达到口感需求。目前,食品中甘草酸、甘草次酸检测参考依据是SN/T 3854—2014《出口食品中天然甜味剂甜菊糖苷、甜菊双糖苷、甘草酸、甘草次酸的测定》[11],该方法前处理繁杂,且检出限高。报道显示,白酒中检出的甜味剂往往是少量,甚至微量,使用现行标准方法难以满足行业的检测需求。目前关于甘草酸、甘草次酸的研究大多在其药理学及临床学等领域,有关含量检测的相关报道较少。而白酒中甜味剂检测方法的研究大多集中于高效液相色谱-质谱联用法(high performance liquid chromatography-mass spectrometry,GC-MS)[12-14],该方法能够实现多种甜味剂的联测,可达到较低的检出限,但天然甜味剂的检出限也通常高于人工甜味剂[1],与市场实际添加情况相悖。液质联用法具有强大的定性功能,但由于样品前处理多采用样液直接或稀释后上机,不可避免产生基质效应,因此在定量上不如高效液相色谱准确,同时,液质联用法对操作人员要求较高,不利于方法的推广。因此,开发一种简单、快速、准确的方法测定白酒中天然甜味剂成为目前重要的一个研究方向。
漂浮有机液滴凝固液相微萃取(liquid phase microextraction based on solidification of floating organic drop,SFODLPME)是近几年液相微萃取技术中发展的比较好的一个分支[15-16],该方法采用小体积有机试剂作为萃取剂,对萃取供相中的分析物进行富集,而后利用萃取剂密度比水小且溶点接近室温的特性达到相分离。方法绿色环保,方便快捷,具有卓越的富集能力,特别适用于痕量物质的提取分离,目前已在食品安全检测领域得到应用研究[17-20]。本研究将SFOD-LPME应用于白酒中甘草酸、甘草次酸的富集,并联合高效液相色谱(HPLC)技术进行测定,为白酒中甜味剂检测方法的建立及优化提供技术参考。
1 材料与方法
1.1 材料与试剂
甘草酸(纯度98%):成都艾科达化学试剂有限公司;甘草次酸(纯度97%):百灵威科技有限公司;正十一醇(纯度>99%)、正十二醇(纯度>98%)、1,10-二氯葵烷(纯度≥99.5%):上海皓鸿生物医药科技有限公司;丙酮、四氢呋喃、甲醇、乙腈(均为色谱纯)、乙醇、盐酸(均为分析纯):国药集团化学试剂有限公司;除特殊说明外,实验用水均为超纯水;白酒样品:随机采购于各大商超及零售店。
1.2 仪器与设备
WatersARCHPLC超高效液相色谱仪(配备二极管阵列检测器):美国Waters公司;RD-C18色谱柱(250 mm×4.6 mm,5 μm):中谱科技有限公司;HWS-26电热恒温水浴锅:上海一恒科学仪器有限公司;HJ-6A多头恒温磁力搅拌器:国华(常州)仪器制造有限公司。
1.3 方法
1.3.1 标准溶液配制
分别准确称取10 mg甘草酸、甘草次酸标准物质于容量瓶中,用甲醇稀释定容至10 mL,得甘草酸、甘草次酸标准储备液,于-18 ℃冷冻保存;根据需要将标准储备液用水稀释至一定浓度作为标准工作液,现用现配。
1.3.2 样品处理
准确移取2 mL白酒置于50 mL玻璃比色管中,于95 ℃水浴10 min,加水溶液20 mL至比色管中进行复溶,涡旋混匀,作为待萃取样品工作液(即萃取供相)。
SFOD-LPME条件:依次在样品工作液加入100 μL 1∶1盐酸溶液(供相溶液pH值约为3)、1.0 g氯化钠、50 μL四氢呋喃以及40 μL正十二醇,将比色管置于多头恒温磁力搅拌器中,在40 ℃搅拌萃取15 min,后停止搅拌,待萃取剂在液面上汇聚成滴,将比色管转移置于冰浴中,待正十二醇凝固后取出,室温下融化,准确吸取30 μL萃取剂加入30 μL甲醇混匀后注入色谱系统。
另取20 mL标准工作液进行相同的SFOD-LPME萃取过程。
1.3.3 色谱条件
流动相:A相为0.02 mol/L磷酸水溶液,B相为乙腈;梯度洗脱程序见表1;流速:1 mL/min;进样量:10 μL,柱温:35 ℃,检测波长:254 nm。
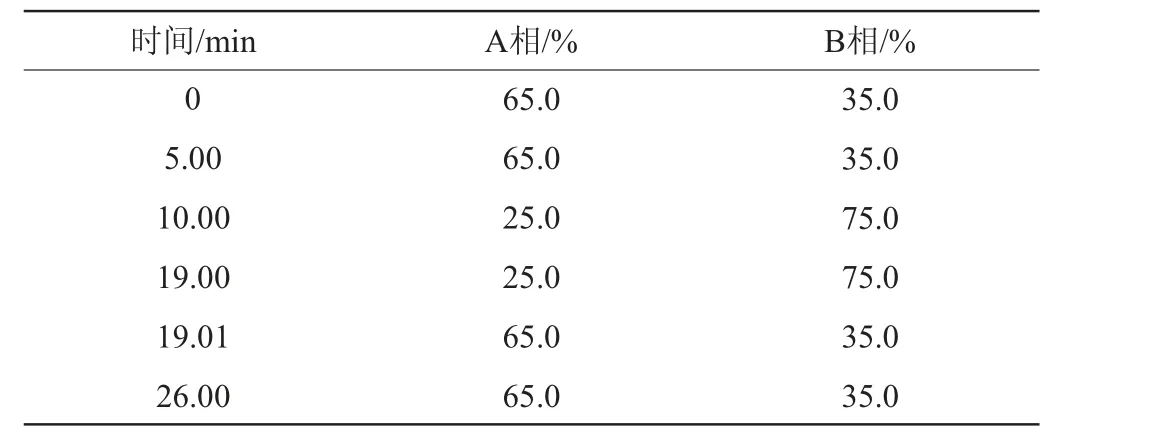
表1 液相色谱梯度洗脱程序Table 1 Gradient elution program of liquid chromatography
1.3.4 萃取条件优化
取20 mL水溶液作为萃取供相,采用单因素控制变量法,分别考察萃取剂(正十二醇、正十一醇以及1,10-二氯癸烷)、萃取剂体积(30~80 μL)、分散剂(四氢呋喃、乙腈、甲醇、丙酮)、分散剂体积(0~600 μL)、供相溶液中盐酸添加量(0~250 μL)、氯化钠添加量(0~2.0 g)、萃取温度(26~60 ℃)、萃取时间(5~30 min)对萃取效率的影响,萃取完成后注入色谱系统进行测定,记录峰面积,以峰面积大小作为萃取效率高低的判断标准,用来确定最佳SFODLPME萃取条件。
1.3.5 方法学评价
(1)标准曲线回归方程、检出限及定量限
分别取适量甘草酸、甘草次酸标准储备液,用水配制成系列质量浓度标准工作液,而后使用最佳的SFOD-LPME条件进行萃取,注入色谱系统进行分析,得到甘草酸、甘草次酸色谱图及峰面积,以标准溶液质量浓度(x)为横坐标,峰面积(y)为纵坐标,制作标准曲线;注入样品萃取空白,计算信噪比,分别以3倍信噪比及10倍信噪比对应的被测物的浓度值,计算得该方法的检出限及定量限。
(2)方法精密度及回收率考察
选取4种不同香型的白酒样品作为加标基质,进行低、中、高3水平加标试验,采用本方法萃取后进行测定,计算精密度试验结果相对标准偏差(relative standard deviation,RSD)及平均回收率,进而完成该方法精密度及准确度考察。
1.3.6 数据分析
采用Empower色谱工作站进行定量分析,Microsoft Excel 2016 统计分析处理数据,Origin 9.5进行图谱处理。
2 结果与分析
2.1 萃取条件优化
2.1.1 萃取剂的选择
SFOD-LPME要求萃取剂密度小于水,并且溶点接近室温,实验考察了满足条件的正十二醇、正十一醇以及1,10-二氯癸烷三种试剂对萃取效率的影响。萃取过程显示,冰浴后,在萃取剂凝固状态的完整性上,正十二醇优于正十一醇及1,10-二氯癸烷,冰浴凝固速度快,操作方便;而1,10-二氯癸烷萃取完成后,液体略呈白色浑浊状态,萃取剂回收较差,可能由于其密度与水最为接近,导致部分滞留于样品溶液中,与水相分离不完全;且色谱图结果显示,使用正十一醇萃取后,目标峰型展宽较大,疑存在萃取干扰。因此选择最佳萃取剂为正十二醇。
在液相微萃取中,两相间的传质系数随着样品溶液体积和萃取剂体积的减少而增大[21],本研究在操作可行的条件下控制样品溶液体积即萃取供相体积为20 mL,并在此体积下进行萃取剂体积的优化,取30~80 μL萃取剂进行萃取效率的考察,考察萃取剂对富集效果的影响。试验结果显示,萃取剂体积越大,相应富集倍数越小,当体积为30 μL时,萃取结果的平行性较差,萃取剂回收重复性低,当体积达到40 μL时,萃取剂回收情况开始呈现稳定状态,综合考虑,确定40 μL正十二醇作为萃取剂。
2.1.2 分散剂的选择
分散剂一般选择既溶于供相,又溶于萃取相的试剂,可以很好的将互不相溶的两相进行充分的混合,在液相微萃取中起着至关重要的作用。该研究选取了四氢呋喃、乙腈、甲醇、丙酮四种溶剂为分散剂,以相同的添加量(100 μL)进行测试,并以不加分散剂为空白试验,结果见图1。选出最佳分散剂后,考察分散剂体积对萃取效果的影响,结果见图2。
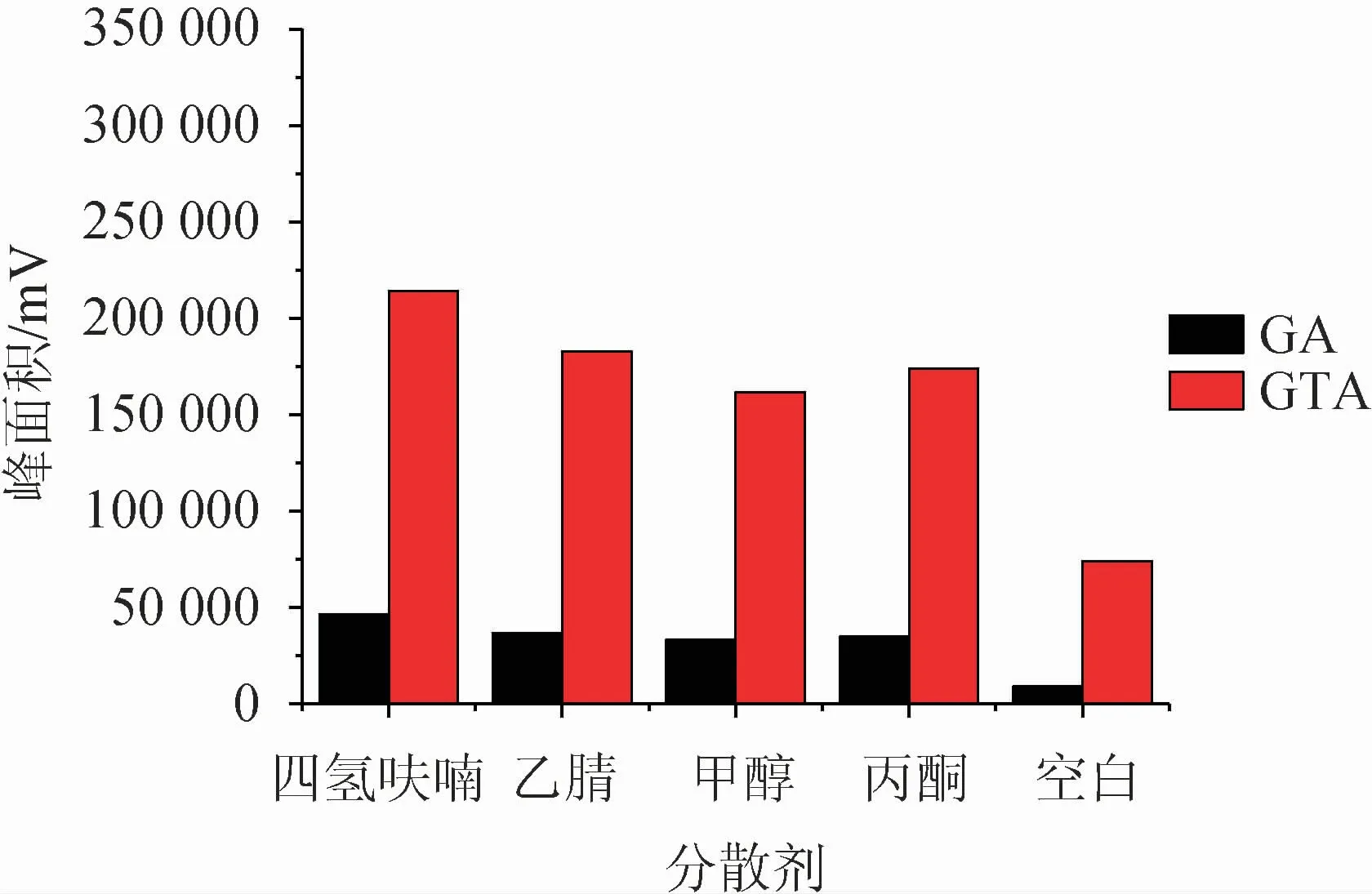
图1 分散剂种类对萃取效果的影响Fig.1 Effect of dispersant varieties on extraction efficiency
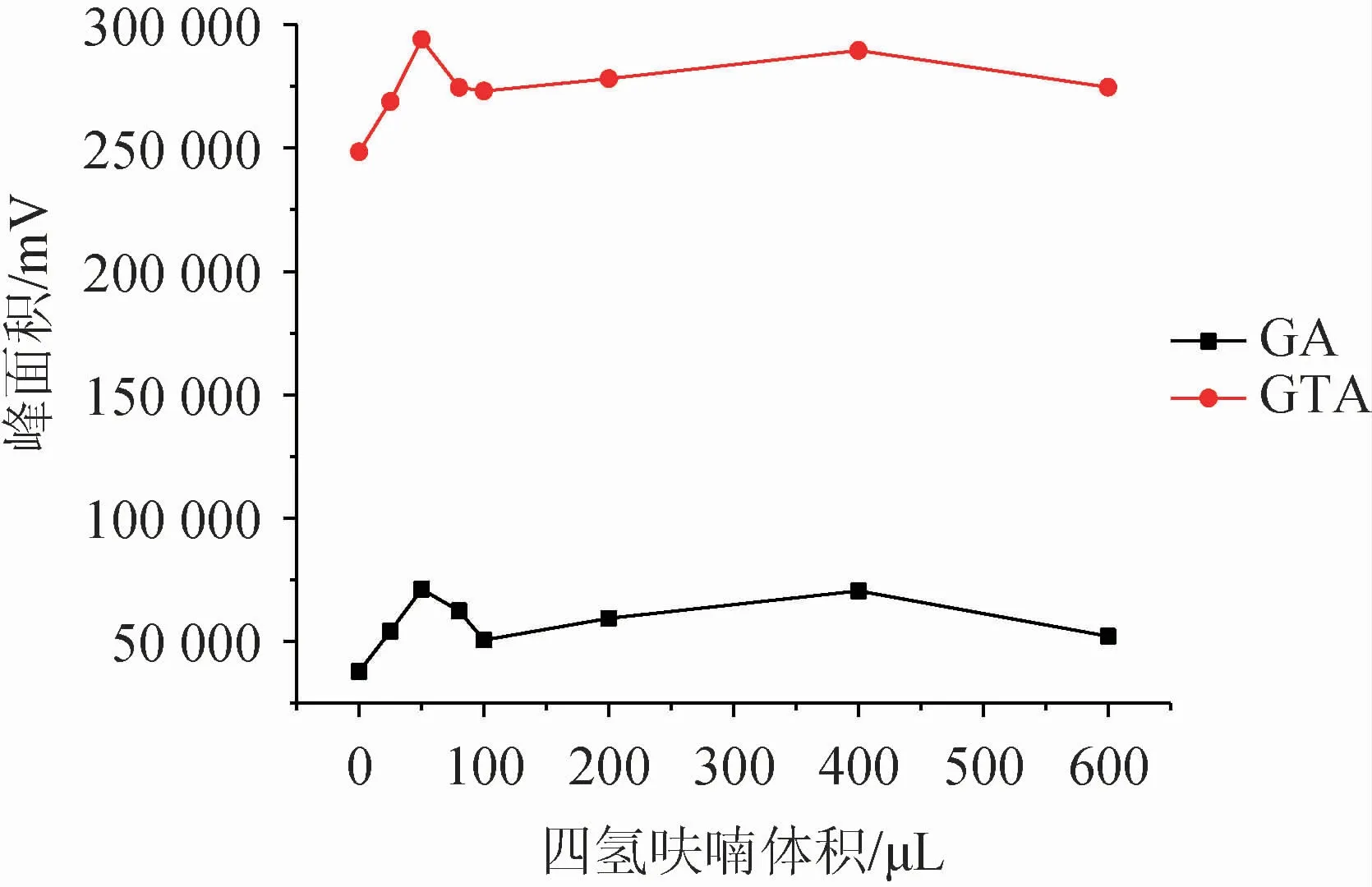
图2 分散剂体积对萃取效果的影响Fig.2 Effect of dispersant volume on extraction efficiency
由图1可知,在不加分散剂的情况下,目标物萃取效果明显很差;而四种溶剂对甘草酸的萃取影响无明显差异,对甘草次酸的影响较大,其中四氢呋喃对该方法体现出较好的适配性。因此选择最佳分散剂为四氢呋喃。由图2可知,随着四氢呋喃添加量的增大,萃取效率逐渐增大,在分散剂体积为50 μL时,萃取效果最佳,随之下降,继续增加四氢呋喃添加量,萃取效果稍有增加但整体趋势趋于平缓。综合考虑,最终确定最佳分散剂为50 μL四氢呋喃。
2.1.3 盐酸添加量的选择
甘草酸由两分子葡萄糖醛酸和一分子甘草次酸组成,和甘草次酸一样,水溶液均呈弱酸性。因此,方法保持供相溶液为酸性体系,使目标化合物呈分子状态,得以快速转移进入萃取相中。该研究以20 mL水溶液为供相体系,考察了1∶1盐酸水溶液的添加量(0~250 μL)对萃取效率的影响,结果见图3。由图3可知,不进行供相溶液酸度控制时,萃取后甘草酸几乎无响应,说明甘草酸完全没有被富集;随着盐酸添加量的增加,萃取效率逐渐增大,到一定程度时,则呈下降趋势。当添加量为100 μL时,目标化合物富集效果最好,此后随盐酸添加量增大而降低,其中甘草次酸降低幅度较大。故确定盐酸添加量为100 μL。
吉林省、四川省、浙江省等出台的孤儿救助的社会政策也对孤儿教育做了较为明确的规定。通过梳理相关政策可以发现,对处于义务教育阶段的孤儿,中央及各地方的教育保障政策都很明确,即实施免费义务教育。而对在普通高中、中等职业学校、高等职业学校和普通本科高校就读的孤儿,除了北京提出免收学费、住宿费、服务性费用,天津提出免收学杂费外,中央和其他地方只是提出将这部分孤儿优先纳入资助政策体系。对于成年孤儿在校就读的,中央和地方文件都只有原则性的规定,即“孤儿成年后仍在校就读的,继续享有相应政策”。由此可见,对于大龄孤儿或者成年孤儿的教育救助或保障政策还需要进一步细化。

图3 盐酸添加量对萃取效果的影响Fig.3 Effect of hydrochloric acid addition on extraction efficiency
2.1.4 氯化钠添加量的选择
在供相溶液中加入一定量的盐,可有效抑制目标化合物的离子化过程,从而增大富集效应。该研究比较了20 mL水溶液为供相体系下氯化钠添加量为0~2.0 g情况下的萃取效果,结果见图4。由图4可知,随着氯化钠添加量的增加,萃取效率有一定改善,当添加量为1.0 g时,甘草酸和甘草次酸均得到了较好的响应,说明此时的盐析效应最有利于萃取的进行,而随着添加量的增大,萃取效率逐渐下降,可能是离子强度过强导致盐离子在萃取剂中产生了静电效应阻止了目标化合物进入萃取剂中[22]。故选择氯化钠添加量为1.0 g。
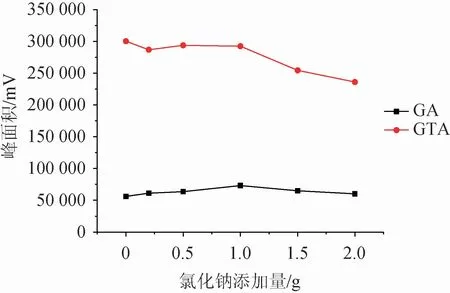
图4 氯化钠添加量对萃取效果的影响Fig.4 Effect of sodium chloride addition on extraction efficiency
2.1.5 萃取温度的选择
温度提高能够加快萃取体系中目标化合物的转移,加速达到萃取平衡。实验从室温(26 ℃)开始,逐渐升高体系温度,考察萃取温度(26~60 ℃)对甘草酸、甘草次酸富集效率的影响,结果见图5。由图5可知,温度对目标化合物萃取效率影响较大,响应值随着温度增高明显呈上升趋势。萃取温度40 ℃时,目标化合物均得到最佳响应,而后下降,疑为温度过高导致萃取剂挥发损失,回收率受影响,正十二醇萃取后液滴表面积随温度升高而增大,到达60 ℃时已呈平铺状态,不利于液滴的分离,实验重复性呈下降趋势。故确定萃取温度为40 ℃。
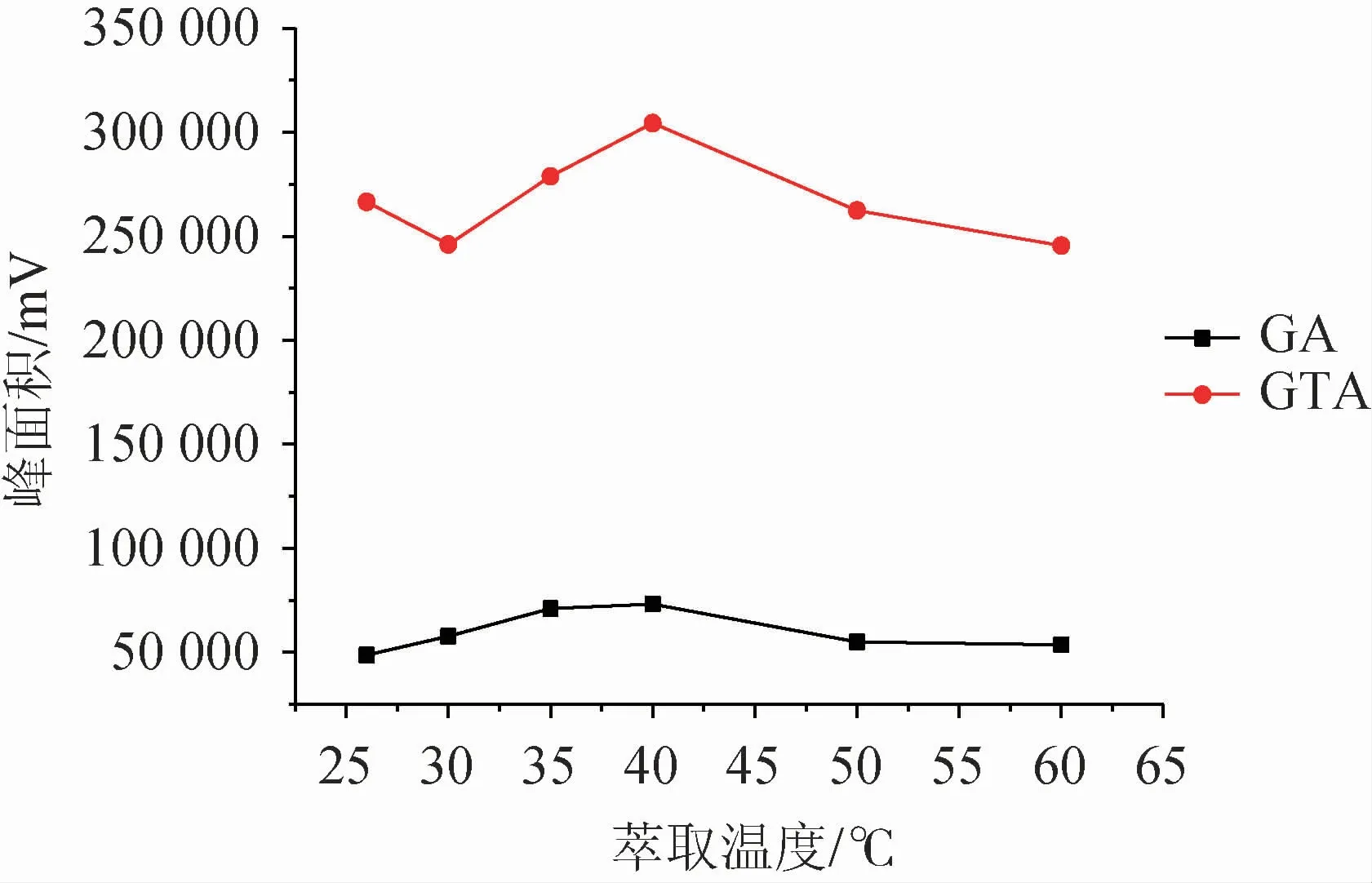
图5 萃取温度对萃取效率的影响Fig.5 Effect of extraction temperature on extraction efficiency
2.1.6 萃取时间的选择
液相微萃取是目标化合物在供相和萃取相之间达到分配平衡的过程,因此对于萃取时间的控制尤为重要。时间过短,萃取未到达平衡,导致萃取不佳;时间过长,同样也会引起萃取剂的损失。实验考察了萃取时间5~30 min内对萃取效果的影响,结果见图6。由图6可知,随着萃取时间的增加,萃取效率先增大后降低。当萃取时间为15 min时,甘草酸达到最佳萃取效果;甘草次酸萃取效果在20 min时达到最佳,但在15 min时也达到了较高水平,考虑到整体实验效率,尽可能在较短的时间完成萃取,最终确定萃取时间为15 min。
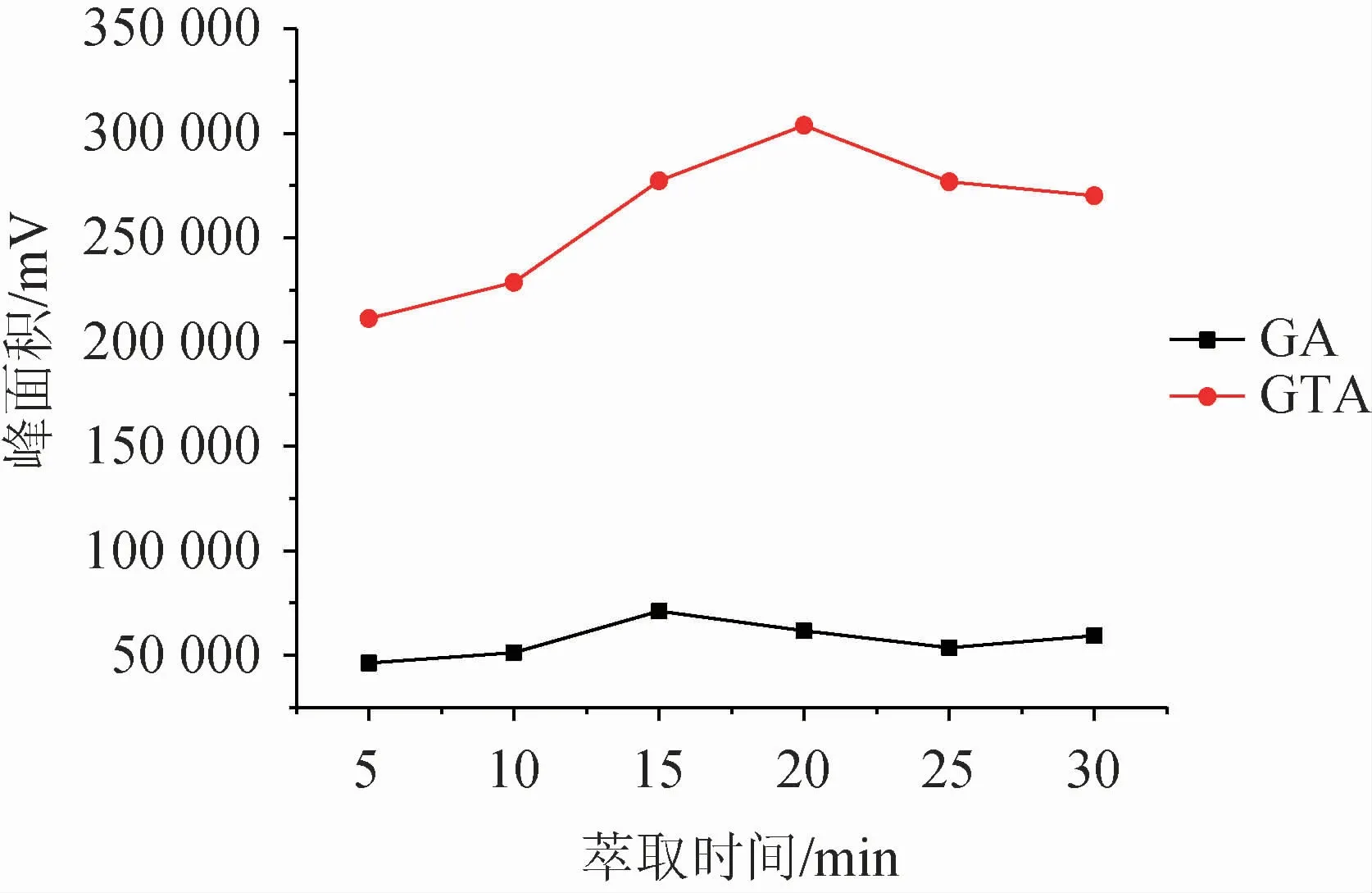
图6 萃取时间对萃取效果的影响Fig.6 Effect of extraction time on extraction efficiency
2.1.7 验证试验
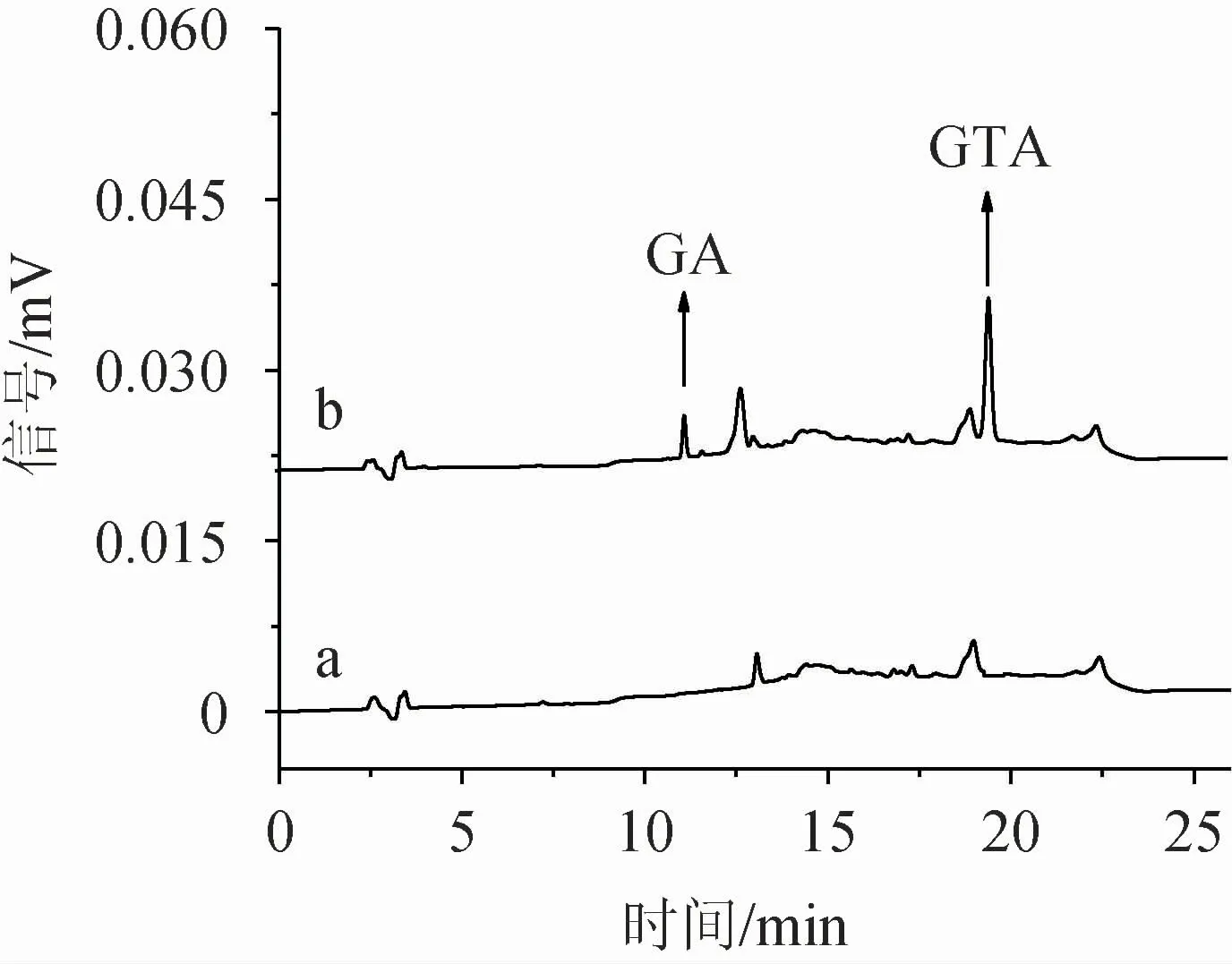
图7 标准溶液及样品中甘草酸及甘草次酸色谱图Fig.7 Chromatograms of glycyrrhizic acid and glycyrrhetinic acid of standard solution and sample
2.2 方法学考察
2.2.1 标准曲线、检出限及定量限
分别取适量甘草酸、甘草次酸标准储备液,用水配制成质量浓度为2.50 ng/mL、5.00 ng/mL、10.0 ng/mL、25.0 ng/mL、50.0 ng/mL的标准工作液,而后使用最佳的SFOD-LPME条件进行萃取,并注入色谱系统进行分析。使用甘草酸、甘草次酸工作液萃取后峰面积(y)与质量浓度(x)绘制标准曲线,结果见表2。
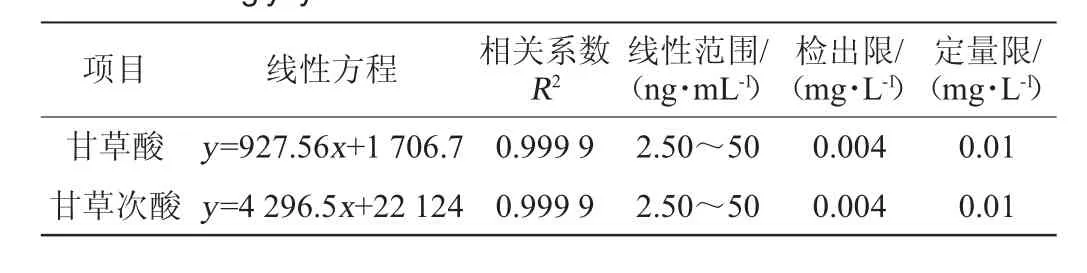
表2 甘草酸和甘草次酸的回归方程、相关系数、线性范围、检测限及定量限Table 2 Regression equation, correlation coefficients, linear ranges,detection limits and quantitation limits of glycyrrhizic acid and glycyrrhetinic acid
由表2可知,在2.50~50.0 ng/mL范围内,甘草酸、甘草次酸显示良好的线性关系,相关系数R2均为0.999 9。注入样品萃取空白,计算信噪比,分别以3倍信噪比及10倍信噪比计算得该方法的检出限及定量限,得甘草酸、甘草次酸检出限为0.004 mg/L,定量限为0.01 mg/L,满足检测要求。
2.2.2 方法精密度及回收率考察
方法选取清香型、浓香型、酱香型、米香型四种风味的白酒基质进行加标回收试验,加标量分别为0.01 mg/L、0.05 mg/L、0.40 mg/L的3水平3平行试验,按照本方法进行萃取分析,加标回收率试验结果见表3。
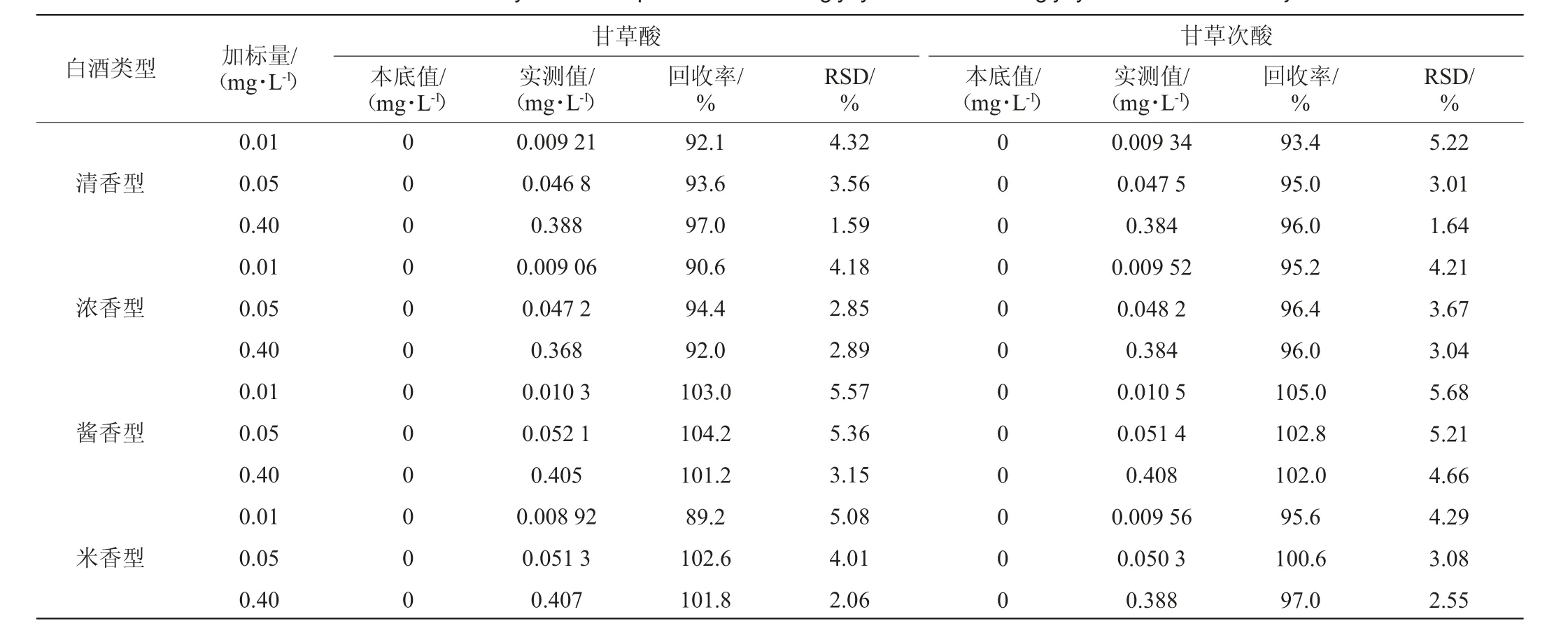
表3 白酒中甘草酸、甘草次酸的加标回收率及精密度试验结果Table 3 Standard recovery rates and precision tests of glycyrrhizic acid and glycyrrhetinic acid in Baijiu
由表3可知,清香型白酒回收率在92.1%~97.0%之间,精密度试验结果RSD在1.59%~5.22%之间;浓香型白酒回收率在90.6%~96.4%之间,精密度试验结果RSD在2.85%~4.21%之间;酱香型白酒回收率在101.2%~105.0%之间,精密度试验结果RSD在3.15%~5.68%之间;米香型白酒回收率在89.2%~102.6%之间,精密度试验结果RSD在2.06%~5.08%之间,四种香型白酒基质间加标回收结果均良好,且无明显差别,说明该方法整体适应性较好,受基质影响小。总体而言,甘草酸加标回收率在89.2%~104.2%之间,甘草次酸加标回收率在93.4%~105.0%之间,精密度试验结果RSD分别在1.59%~5.57%及1.64%~5.68%之间,精密度和回收率均符合检测要求。
2.3 实际样品的测定
对随机采购的四种香型共50份白酒样品进行检测,每份样品平行测定3次,检测结果显示,样品中均无甘草酸、甘草次酸检出。
3 结论
随着市场对天然甜味剂的认可,其替代传统甜味剂必然是大势所趋,虽然甘草酸、甘草次酸作为甜味剂添加在食品中不仅能够改善口感,还具有营养保健的作用,但是对于白酒行业来说,这是对传统工艺的伤害,更是对消费者的欺瞒,甚至会引发企业间不正当竞争。因此,对白酒中天然甜味剂含量展开监测仍然十分必要。高效天然甜味剂的推广和使用,将对检测方法的灵敏度提出更高的要求,同时也是对白酒中甜味剂违法添加的监管提出了一个新的挑战。本研究建立SFOD-LPME-HPLC法测定白酒中天然甜味剂甘草酸、甘草次酸,方法灵敏度高,操作简单,富集效果显著,适用于白酒中微量甘草酸、甘草次酸的分析检测,可为白酒中添加剂的检测提供方法参考,有助于建立相关标准及预警机制,对市场形成更有效的监管。
