饲用大豆全株蛋白含量关联SSR 标记筛选
2023-09-06李忠旺陈光荣刘新星杨如萍朱天地陈子萱陈玉梁
李忠旺,陈光荣,刘新星,杨如萍,朱天地,陈 琛,陈子萱,陈玉梁
(1.甘肃省农业科学院生物技术研究所, 甘肃 兰州 730020;2.甘肃省农业科学院旱地农业研究所, 甘肃 兰州 730070)
大豆(Glycine max)起源于中国,栽培历史悠久,其籽粒不仅为人类提供粮油,更是动物最主要的植物蛋白来源。栽培大豆籽粒收获后的成熟秸秆由于木质化程度高、粗蛋白含量低而不能广泛用作动物饲料[1]。近年来利用野生大豆和栽培大豆杂交培育的饲用大豆是以收获植株营养体为主的新型品种,具有生长势旺盛、适宜性广、蛋白含量高、抗逆性强等特点,作为优质的一年生饲草还兼有鲜草产量高、营养丰富、适口性好、草食畜均喜采食等优点,可作为优良的草食畜青饲料[2-4]。因此,选育全株高蛋白含量的饲用大豆新品种具有极其重要的现实意义。然而,由于饲用大豆选育一般都是以栽培大豆和野生大豆为原始双亲,需经过多轮回交转育的过程,且全株蛋白含量为受多基因调控的数量性状,性状鉴定需要在生长晚期,完全破坏植株后才能鉴定,鉴定难度大、费用高。这给全株高蛋白含量饲用大豆选育工作带来了极大的麻烦。鉴于此,探索应用分子标记辅助选择技术开展高蛋白含量饲用大豆新品种选育,是解决难题的有效手段。
微卫星序列SSRs (simple sequence repeats)作为一种多态性高、重复性好且较稳定的分子标记,常用于遗传多样性研究,由于其兼具共显性、易扩增、基因组内分布广泛等优点,同时也是关联分析研究理想的分子标记[5]。前期大量研究工作都是利用双亲 构 建 的 重 组 自 交 系(recombinant inbred line, RIL)分离群体定位与大豆籽粒蛋白含量相关的SSR 标记,这些SSR 标记大部分是在单一分离群体中被定位,缺少在核心种质自然群体中的验证,限制了其在育种实践中的应用价值。关联分析是利用等位基因间的连锁不平衡(linkage disequilibrium, LD)关系进行性状与标记间的相关性分析,可检测自然群体中与性状相关的基因位点,进而应用于分子标记辅助选育工作中[6]。
本研究以甘肃省农业科学院大豆育种课题组前期筛选的55 份饲用大豆核心种质自然群体为受试材料,利用前人定位筛选的40 个大豆籽粒蛋白含量表型遗传解释率高的SSR 标记,采用关联分析的方法,初步筛选在自然育种群体中与大豆全株蛋白含量显著关联的SSR 标记,为进一步应用分子标记辅助选育高蛋白含量饲用大豆新品种提供理论依据。
1 材料与方法
1.1 供试材料
本研究选取甘肃省农业科学院大豆育种课题组前期筛选的适宜于饲用大豆选育的55 份核心种质资源作为供试群体材料,其中野生大豆资源7 份,栽培大豆资源34 份,半野生牧草大豆资源3 份,野生大豆与栽培大豆杂交后代高代品系11 份。2020年和2021 年分别将供试大豆品种(系)种植于甘肃省农业科学院黄羊试验场,采用随机区组设计,3 次重复,行长3.0 m,行距0.5 m,株距0.1 m。采样时选择小区中间株去除边际效应,每个品种每个重复小区选择3 株作为3 个生物学重复测定全株粗蛋白含量。
1.2 SSR 标记筛选及引物设计
采用文献检索法,收集国内外文献报道的大豆籽粒蛋白含量关联SSR 分子标记,从中筛选出解释率较高的连锁SSR 标记40 个作为目标对象,并从SoyBase 数据库(https://www.soybase.org/)查找这些标记的引物序列,委托上海生工生物工程技术服务有限公司合成引物备用。检索到的SSR 标记信息如表1 所列。
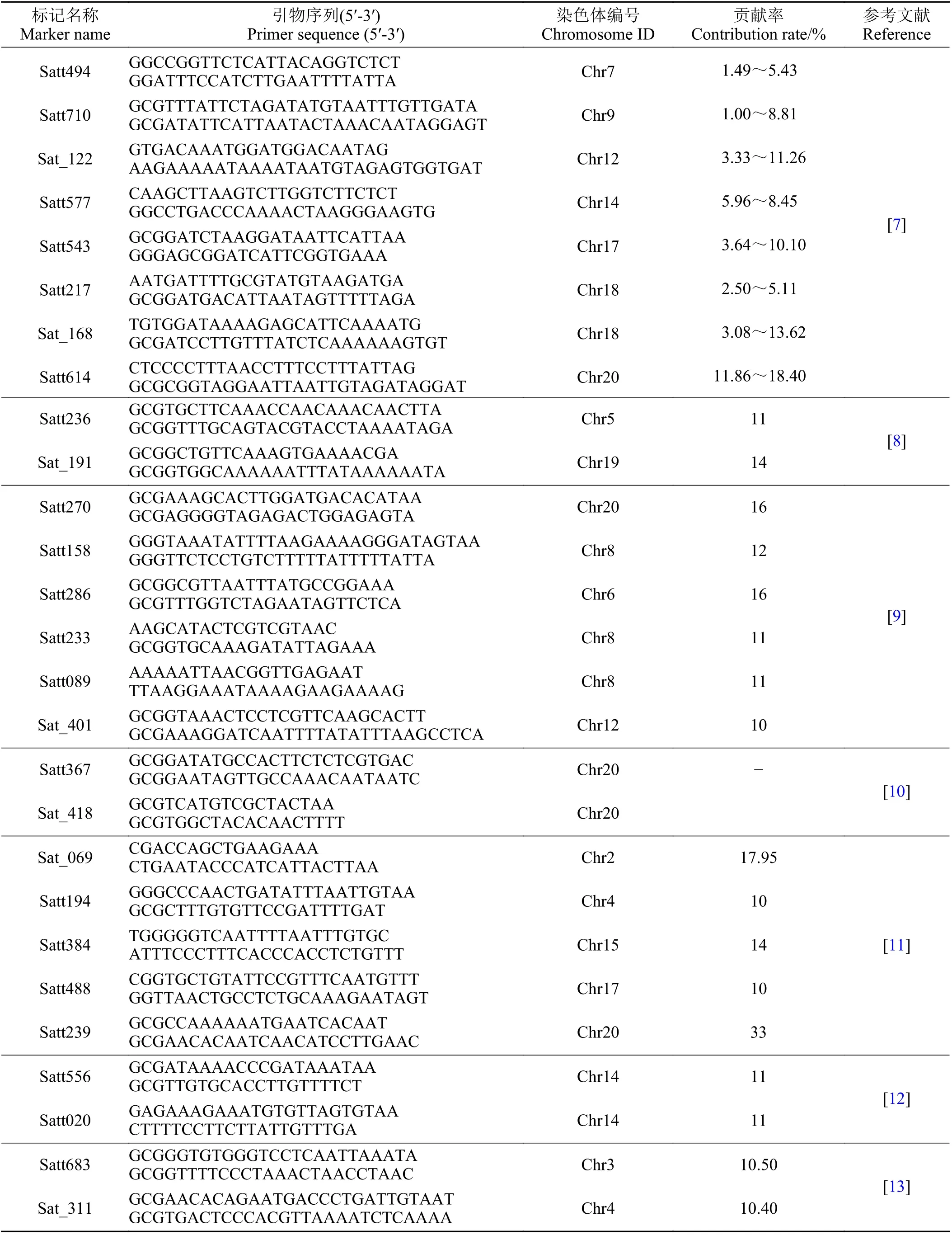
表1 大豆蛋白含量相关SSR 标记信息列表Table 1 List of SSR marker information related to soybean protein content
1.3 大豆全株蛋白含量测定
在大豆鼓粒期,各受试材料每个重复小区内选择连续3 个单株作为全株蛋白含量测定样品,记录并称其鲜重后带回实验室于60 ℃干燥箱中烘干至恒重,将烘干的大豆粉碎后用于测定植株含氮量(采用凯氏定氮法),测定方法参照《土壤农化分析》[18],植株粗蛋白含量 = 植株氮含量 × 6.25。
1.4 SSR 标记检测
供试材料新鲜叶片经液氮研磨后,每个样品取约100 mg,使用杭州倍沃医学科技有限公司生产的植物基因组DNA 提取试剂盒进行样品DNA 的提取,提取完成后用超微量分光光度计测定其浓度和纯度,并根据所测数据将DNA 稀释至100 ng·μL-1,4 ℃保存备用。PCR 扩增体系总反应体积20 μL,包括2 × Taq Master Mix 10 μL,DNase-Free Water 7 μL,Forward Primer 1 μL,Reverse Primer 1 μL,模板DNA 1 μL。反应程序:95 ℃热启动3 min,95 ℃变性45 s,48~65 ℃退火30 s,72 ℃延伸1 min,30 个循环后,72 ℃终延伸5 min 后结束。PCR 扩增产物用8%的非变性聚丙烯酰胺凝胶电泳分离,银染显色后拍照留存用于数据统计。
1.5 数据统计与分析
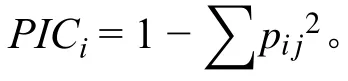
采用Structure 2.3.4 软件进行群体遗传结构分析,估计最佳群体组群数K,取值范围为1~10,每个K 值重复运行3 次,参照Evanno 等[19]提出的ΔK 值方法确定合适的K 值,并计算Q 参数。利用NTSYS 2.10 软件计算遗传相似系数和遗传距离,并进行聚类分析。运用TASSEL2.1 软件一般混合线性模型(general linear model, GLM)和混合线性模型(mixed linear model, MLM),结合蛋白质含量与分子标记数据,进行SSR 标记和全株蛋白含量的关联分析,利用位点显著性标准P< 0.05 且贡献率大于1%对关联位点进行判定,筛选与大豆全株蛋白含量相关联的SSR 标记位点。
2 结果分析
2.1 供试材料全株蛋白含量检测
将55 份饲用大豆核心种质资源2020 年和2021年测得的鼓粒期全株蛋白含量平均值作为表型参数。从测试结果来看,55 份资源中蛋白含量最高的为‘汾豆105’(24.8%),最低的为高代品系‘XZY8’(14.9%);野生大豆全株蛋白含量相对较高,栽培大豆全株蛋白含量相对较低(表2)。
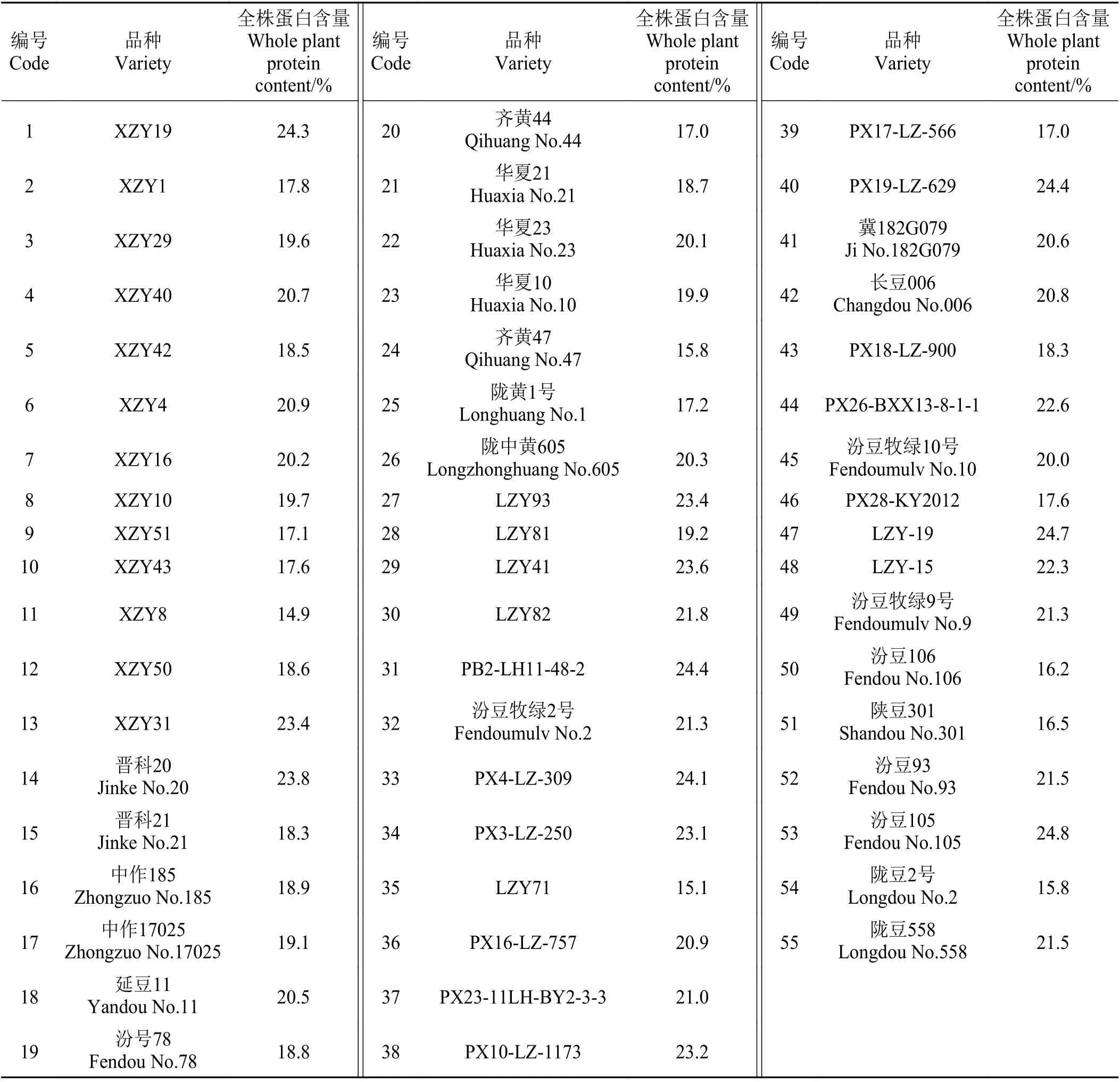
表2 参试品种名称及全株粗蛋白含量测定结果Table 2 Name of tested varieties and determination results of whole-plant protein content
2.2 基于SSR 标记的遗传多样性分析
从40 个SSR 标记中最终筛选出扩增条带清晰、重复性高的SSR 标记21 个,21 个标记在55 份大豆材料中共检测出118 个等位变异,每个标记的等位基因数变幅为3~10 个,平均为5.6 个。其中,多态性位点最丰富的是Sat_100 标记,Sat_168、Satt233 和Satt077 标记检测到的等位变异较少,分别为3、4 和4 个。有效等位基因数(Ne)变幅为1.662 5~6.768 7,平均为3.049 4;Shannon’s 信息指数变幅为0.810 0 (Satt384)~2.038 4 (Sat_100),平均为1.279 2;多态信息含量(PIC)变幅为0.243 6 (Satt126)~0.970 4 (Satt494),平均为0.635 8 (表3)。PIC 值大于平均数占比为52.38%,表明本研究筛选出的标记基因多样性较高。图1 为引物Satt020 在55 份大豆核心种质供试材料中的扩增图谱。
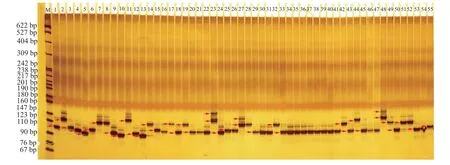
图1 55 个大豆品种的Satt020 引物PCR 扩增图谱Figure 1 PCR products fingerprints of soybean varieties using primer Satt020
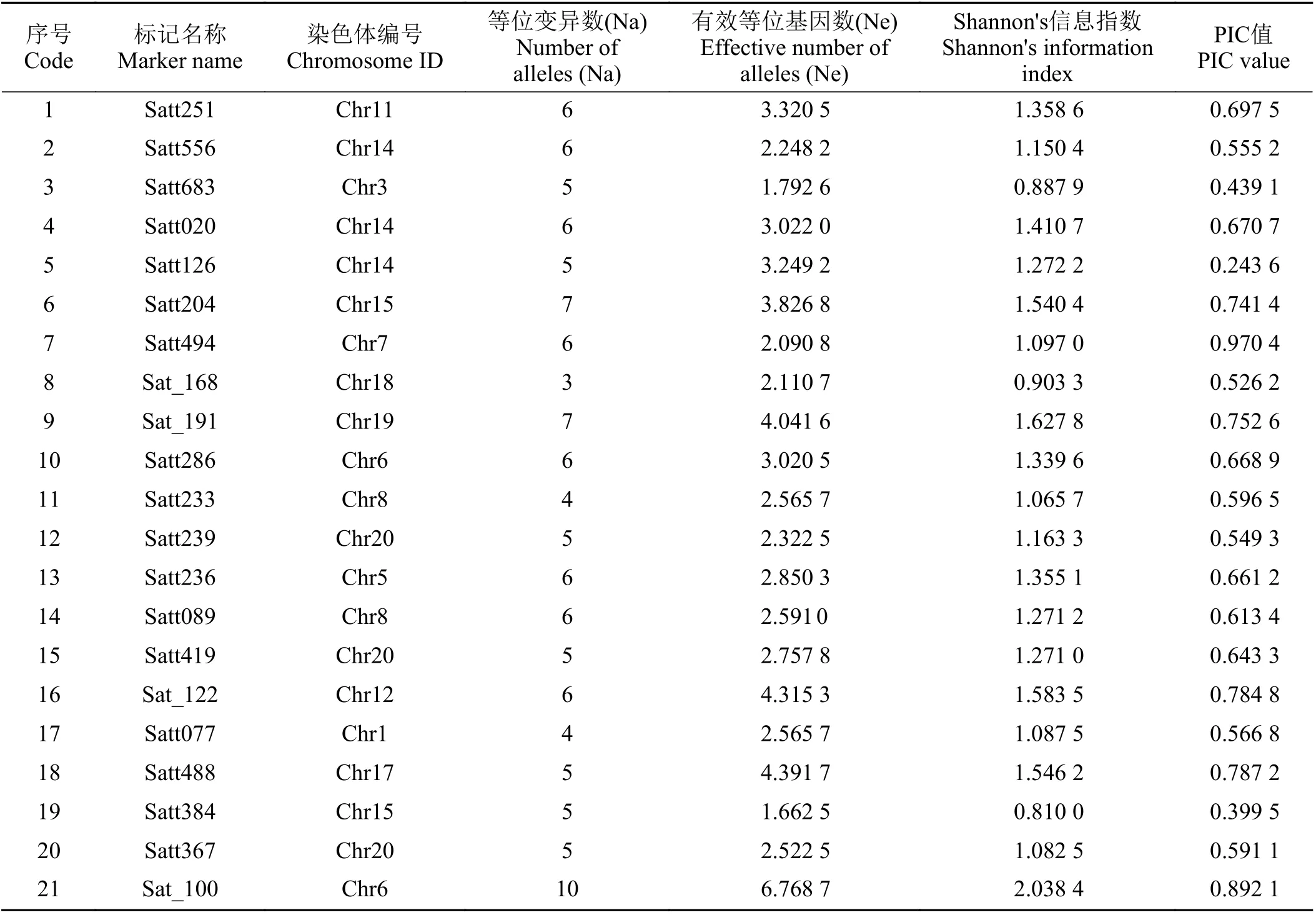
表3 基于21 个SSR 标记的55 份大豆种质的遗传多样性参数Table 3 Genetic diversity indicators of 55 soybean based on 21 pairs of SSR primers
2.3 聚类分析
根据NTSYS 2.10 软件计算,55 份供试大豆种质材料间的遗传相似度变化范围为0.621 8~0.958 0,平均为0.768 1。利用NTSYS 软件的SHAN 程序进行UPGMA 聚类分析,在遗传距离等于0.74 处,供试的55 份大豆种质可以分为3 组。第Ⅰ组包括6 份大豆材料,第Ⅱ组包括48 份大豆材料,第Ⅲ组包括1 份大豆种质,聚类分组结果并没有体现出大豆的品种类型区分(图2)。
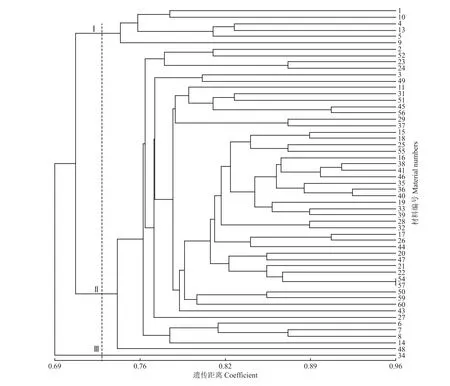
图2 供试大豆种质的UPGMA 聚类图Figure 2 UPGMA dengrogram of 55 soybean materials
2.4 群体遗传结构分析
利用Structure2.3.4 软件对55 份供试大豆材料进行群体遗传结构分析,计算每份材料的Q 值。随着K 值增大,对数似然函数值lnP (D)持续增大,无明显拐点,所以不能判断亚群数,需要通过ΔK 来进一步确定(图3A)。当K = 4 时,ΔK 值最大,因此可将55 份材料分成4 个亚群(图3B、图3C)。根据个体在亚群中的概率,如果概率大于0.6 则划分到相应的亚群,如果概率小于0.6 则划分到混合类群。结果表明,供试大豆材料中,53 份材料的Q 值不低于0.6,推断这些材料的遗传组成相对单一,占比为96.36%。其中,亚群POP1 包含18 份材料,亚群POP3包含14 份材料,亚群POP4 包含16 份材料,这3 个亚群共包含48 份材料,与聚类分析中的第Ⅱ组材料相似;亚群POP2 包含5 份材料,与聚类分析中第Ⅰ组材料基本一致,剩余的2 份材料在4 个亚群内的Q 值均低于0.6,因此划分为一个混合类群,占比为3.63%,表明这两份材料遗传背景复杂。群体结构分析与聚类分析的结果相比较,二者有较高的相似度,但并不完全一致。
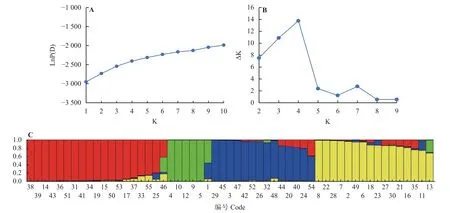
图3 55 份供试大豆材料的群体结构Figure 3 Population structure of 55 soybean materials
2.5 供试大豆蛋白含量与SSR 标记关联分析
以55 份供试大豆材料对应的Q 值作为协变量,利用Tassel 2.1 软件的GLM 和MLM 模型,分析大豆蛋白含量相关联的SSR 标记,并确定其解释率。GLM 检测结果显示,共检测到6 个标记与大豆蛋白含量显著相关(P< 0.05),表型变异的解释率范围为10.36%~16.65%,MLM 分析检测到的相关标记与GLM 分析结果一样,变异解释率的范围为9.38%~16.18%。其中标记Satt_168 与蛋白质含量显示极显著相关(P< 0.01),位于连锁群Chr18 上(表4)。
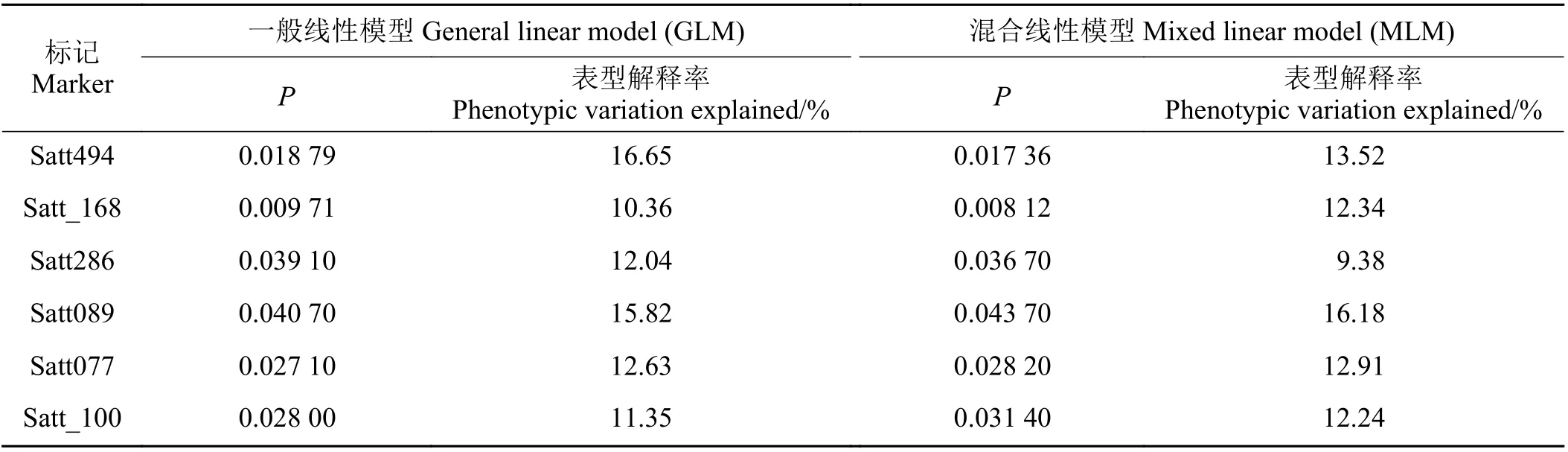
表4 与大豆全株蛋白含量显著关联的SSR 标记Table 4 SSR markers significantly associated with soybean whole plant protein content
3 讨论与结论
大豆因其具有生物固氮功能,所以全株具有较高的蛋白含量,作为饲草可为草食畜提供蛋白质来源,因而早在19 世纪初就被美国作为饲草引进栽培,且很长时间内被作为优质饲草利用[20]。大豆植株蛋白质含量随成熟度的增加而逐渐增加,在鼓粒期收获,其产草量和饲用价值最高[21-22],因此本研究选择在鼓粒期检测饲用大豆选育核心种质资源全株蛋白含量。从检测结果来看,野生型大豆全株蛋白含量相对较高,饲用大豆次之,栽培大豆最低,但兼顾到产草量及适口性则是饲用大豆最优。因此,选择优良的野生大豆和栽培大豆为亲本,选育全株蛋白含量高、产草量高、抗逆性好、综合性状优良的饲用大豆作为一年生饲草应用,具有重要的现实意义。
普遍认为大豆蛋白含量是受多基因控制的数量性状,利用分子标记技术可有效加速数量性状育种进程。已报道的大豆蛋白含量相关分子标记有很多,但是真正可用于育种实践的还比较少。采用关联分析的方法筛选可用的分子标记是一种较好的方法,具有检测等位变异位点多,效率高的特点[23]。本研究以前期筛选的适宜于饲用大豆选育的55 份核心种质资源作为供试群体材料,采用关联分析的方法筛选可应用于育种实践的标记位点。结果显示,40 个SSR 标记中多态性高、重复性好的标记位点有21 个,等位基因数变幅在3~10 个,利用GLM_Q 值 模 型 和MLM_Q + K 值 模 型[24]的 关 联 分析结果一致,共筛选出了6 个与全株蛋白含量相关联的SSR 标记,分布在染色体Chr7、Chr18、Chr6、Chr8、Chr1 和Chr6 上,其中标记Satt_168与大豆蛋白含量在P< 0.01 水平上极显著相关。因此,利用关联分析的方法在已报道的分子标记中筛选可用于育种实践的辅助选择标记。
