凝血功能障碍患儿临床表现、实验室检查及预后分析
2023-08-31路幼佳刘洪军陈天平屈丽君吕文秀
路幼佳, 刘洪军, 陈天平, 屈丽君, 韦 楠, 吕文秀
安徽省儿童医院 血液肿瘤科,安徽 合肥 230000
凝血功能障碍(coagulation disorders,CD)是指由于血管壁异常、凝血因子缺乏、血小板减少、药物因素等导致血液凝固障碍,从而引起出血的一种异常表现[1]。该病患者多表现为鼻出血、牙龈出血、皮下瘀斑、月经量过多等,严重者会出现内脏出血、休克等,严重威胁患者的生命健康。根据病因,CD可分为遗传性CD(血友病、遗传性出血性毛细血管扩张症、血管性假血友病等)和获得性CD(由于凝血因子缺乏、血小板减少、药物因素等导致的出血)[2]。纠正凝血功能异常是CD的治疗目标。遗传性CD一般不可根治,需要终生行凝血因子替代治疗,进而控制病情[3]。获得性CD经过对因治疗后,一般可以根治凝血功能异常,不会影响寿命,若未积极治疗原发疾病导致病情进展,可能并发弥散性血管内凝血、多脏器功能衰竭等,一般不可根治,预后较差[4]。目前,关于儿童CD的发病机制尚未明确。本研究分析总结CD患儿的临床表现、实验室检查、预后等情况,旨在为临床治疗儿童CD提供更多决策依据。现报道如下。
1 对象与方法
1.1 研究对象 选取自2017年7月至2022年6月安徽省儿童医院收治的209例CD患儿为研究对象。其中,男性138例,女性71例;年龄1个月至17岁,中位年龄4岁。纳入标准:符合CD相关诊断标准[5];患儿家属知晓本研究,并签署知情同意书。排除标准:经医师评估预计生存期<3个月;中途退出研究或失访;存在心脑血管疾病;存在精神障碍疾病。本研究符合《赫尔辛基宣言》相关伦理准则。
1.2 治疗方法 遗传性CD患儿均行替代治疗。血友病A患儿输注人凝血因子Ⅷ(华兰生物工程股份有限公司,国药准字S20003004)替代治疗,血友病B患儿输注人凝血酶原复合物(华兰生物工程股份有限公司,国药准字S20083057)替代治疗,因子Ⅶ缺乏症患儿输注注射用重组人凝血因子Ⅶa(丹麦诺和诺德公司,进口药品注册标准S2015006)替代治疗,因子Ⅻ缺乏症和因子Ⅴ缺乏症患儿均输注血浆替代治疗,低纤维蛋白血症患儿输注人纤维蛋白原(fibrinogen,FIB,山东泰邦生物制品有限公司,国药准字S20170007)替代治疗。获得性CD患儿均在治疗原发病的同时,给予血浆、FIB、维生素K1等支持治疗。维生素K依赖性凝血因子缺乏症患儿给予维生素K1注射液(成都倍特药业有限公司,国药准字H32021752)进行治疗;肝源性CD患儿在积极治疗控制原发病的同时,输注血浆支持,同时输注冷沉淀或FIB治疗;免疫性疾病导致的CD患儿在治疗原发病的基础上,输注血浆支持治疗;药物导致的FIB合成减少需要输注冷沉淀或人FIB治疗;无明显诱因的凝血功能异常,通过输注血浆支持治疗。
1.3 观察指标 入院后详细询问病史、家族出血史及临床表现,并进一步完成凝血功能指标[凝血酶原时间(prothrombin time,PT)、部分活化凝血酶原时间(activated partial thromboplastin time,APTT)、FIB]、凝血因子活性、肝功能[血清总胆红素(total bilirubin,TBIL)、天门冬氨酸氨基转移酶(aspartate transaminase,AST)、丙氨酸氨基转移酶(alanine aminotransferase,ALT)]、肾功能(血肌酐、尿素氮、尿酸)、病原学检测[EB病毒、巨细胞病毒、支原体等]、骨髓形态学、白血病免疫分型、染色体、融合基因等相关检测。上述资料均由本院专职人员收集。患儿出院后,每3个月以电话或门诊形式进行随访,以2022年12月为随访终点,记录所有患儿的预后情况。
1.4 统计学方法 采用SPSS 22.0统计学软件处理数据。非正态分布的计量资料以中位数(四分位数间距)[M(P25,P75)]表示,组间比较采用Mann-WhitneyU检验。计数资料以例(百分率)表示,组间比较采用χ2检验。以P<0.05为差异有统计学意义。
2 结果
2.1 患儿病因及原发病 209例CD患儿中,遗传性CD 38例(纳入遗传性CD组),其中,血友病A 24例,血友病B 8例,因子Ⅶ缺乏症2例,因子Ⅺ缺乏症2例,因子Ⅴ缺乏症1例,低FIB血症1例;获得性CD 171例(纳入获得性CD组),其中,维生素K依赖性凝血因子缺乏症11例(5例为溴鼠灵中毒,6例为晚发维生素K缺乏症),25例源于感染或溶血性贫血[包括巨细胞病毒感染(血液巨细胞病毒DNA定量阳性)1例,EB病毒感染(EB病毒DNA定量阳性)2例,支原体感染(支原体IgM抗体阳性)5例,噬血细胞综合征(hemophagocytic lymphohistiocytosis,HLH)15例(其中,EB相关HLH 10例),溶血性贫血2例],5例源于免疫性疾病(包括免疫性血小板减少症1例,过敏性紫癜1例,系统性红斑狼疮3例),124例源于急性淋巴细胞白血病患儿应用培门冬酶化疗时所致的凝血异常,6例无明显诱因单纯性凝血功能异常。
2.2 两组患儿出血情况比较 遗传性CD组出血率为86.84%(33/38),与获得性CD组的78.95%(135/171)比较,差异无统计学意义(P>0.05)。见表1。
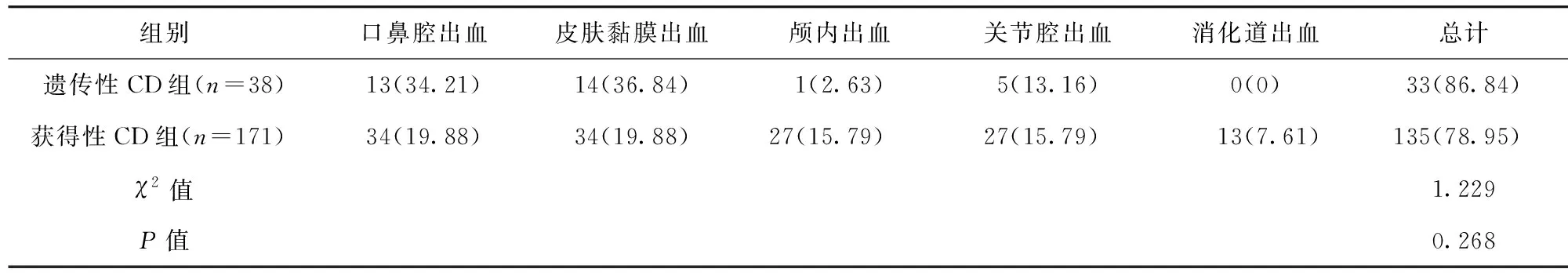
表1 两组患儿出血情况比较/例(百分率/%)
2.3 两组患儿凝血功能指标比较 两组PT比较,差异无统计学意义(P>0.05)。获得性CD组APTT、FIB低于遗传性CD组,差异有统计学意义(P<0.05)。见表2。

表2 两组患儿凝血功能指标比较[M(P25,P75)]
2.4 两组患儿肝功能指标比较 两组AST水平比较,差异无统计学意义(P>0.05)。获得性CD组TBIL、ALT水平高于遗传性CD组,差异有统计学意义(P<0.05)。见表3。

表3 两组患儿肝功能指标比较[M(P25,P75)]
2.5 两组患儿肾功能指标比较 两组患儿血尿素氮、尿酸水平比较,差异无统计学意义(P>0.05)。获得性CD组血肌酐水平高于遗传性CD组,差异有统计学意义(P<0.05)。见表4。

表4 两组患儿肾功能指标比较[M(P25,P75)]
2.6 两组患儿预后情况比较 两组患儿均未出现死亡,预后良好。
3 讨论
CD是较常见的出血性疾病,主要表现为发生轻微外伤后,皮肤关节容易出现血流不止的症状。该病主要是由于患儿骨髓功能病变造成的严重血液系统疾病,主要表现为人体凝血因子、血小板等凝血功能物质降低,造成人体凝血功能异常[6]。临床治疗CD的首要步骤为查明引发出血的原因,根据患者实际病情正确补充凝血因子,提高凝血因子的数量与浓度,达到止血的效果[7]。本研究中,获得性CD患儿与遗传性CD患儿PT无明显差异,而获得性CD患儿APTT、FIB较低。获得性CD患儿常见病因包括特殊药物、各种原因引起的肝功能异常、晚发维生素K缺乏症、风湿免疫疾病等。此外,急性白血病、淋巴瘤化疗过程中应用的培门冬酶也易引起患儿CD[8],其主要原因是抑制蛋白合成,导致FIB、各类凝血因子水平降低。HLH患者发生CD最常见的原因是FIB水平下降[9-10],FIB<1.5 g/L是HLH的诊断指标之一[11]。这提示,FIB水平可能辅助区分获得性CD与遗传性CD。本研究发现,获得性CD患儿与遗传性CD患儿AST、血尿素氮、尿酸水平无明显差异,而获得性CD患儿TBIL、ALT、血肌酐水平高于遗传性CD患儿,分析原因可能为获得性CD多源于急性淋巴细胞白血病,应用培门冬酶进行化疗可致重度脂肪肝伴胆汁淤积,出现凝血异常,肝损伤可能性较高[12]。
本研究中,209例CD患儿均未出现死亡,预后良好。遗传性CD(如血友病)主要采取凝血因子替代治疗,通过补充体内缺乏的凝血因子促进凝血,以达到止血的目的。20世纪50年代末和60年代初,新鲜冰冻血浆或凝血酶原复合物开始用于治疗血友病[13]。同时,应该根据出血程度、所缺乏凝血因子成分及活性等情况进行替代治疗。当血友病患儿发生出血事件时,特别是颅内出血、颈部/咽喉部出血、消化道出血等可能危及生命的严重出血事件,需要立即给予凝血因子替代治疗[14]。本研究中,因子Ⅶ缺乏症2例,因子Ⅺ缺乏症2例,因子Ⅴ缺乏症1例,低FIB血症1例,均在正常生活中无出血倾向,拟进行手术等情况时,输注相应凝血因子制品或血浆防止出血。对于获得性CD,积极治疗原发病是终止凝血异常的关键。维生素K缺乏为多因子联合缺乏,常有自发性皮肤、黏膜和内脏出血倾向,去除病因和维生素K序贯应用可有效治疗。HLH患者体内免疫调节异常,主要为淋巴细胞、单核细胞和巨噬细胞系统异常激活与增殖,分泌大量炎症因子,异常活化的巨噬细胞在细胞因子作用下,刺激单核细胞上尿激酶纤溶酶原激活物受体表达,从而促进纤溶酶原细胞的激活,最终导致纤维蛋白降解[15]。
HLH合并CD的治疗关键为及时给予免疫治疗,清除炎症因子,阻断细胞因子风暴,进而降低死亡风险[16]。此外,输注FIB、凝血酶原复合物、血浆等替代治疗也是治疗过程中不可缺少的一部分。白血病患儿治疗过程中应用的培门冬酶是聚乙二醇与左旋门冬酰胺酶(L-asaraginase,L-ASP)共价连接而成的新型门冬酰胺酶制剂。L-ASP是蛋白质与核酸生物合成所必需的氨基酸,可催化血浆中门冬酰胺水解,耗竭门冬酰胺,抑制蛋白质合成,从而导致纤维蛋白溶酶、FIB、各类凝血因子等水平下降,出现凝血功能不全[17]。当患者FIB处于最低下限水平时,应及时输注FIB或血浆替代治疗,恢复凝血功能。脓毒症、重症肺炎、EB病毒感染等均可引起CD,主要由于感染后,血管内皮细胞损伤,释放大量组织因子进入血液,导致凝血异常[18]。本研究中,6例原因不明的CD患儿以APTT延长为主,多因鼻出血就诊,经过及早控制出血以减少颅内出血风险,补充维生素K和输注血浆纠正CD等处理,后期随访中患儿凝血功能均恢复正常。
综上所述,儿童获得性CD多见,出血部位主要为口鼻腔和皮肤黏膜;遗传性CD需要特异性凝血因子终生替代治疗,预后取决于凝血因子缺乏的程度及干预的积极性;获得性CD需要从根本上治疗原发病,血液制品短期替代治疗,预后主要取决于原发病的治疗效果。
