Menke-Hennekam 综合征表型及基因型分析
2023-08-23唐雅楠叶贤涛顾学范余永国
唐雅楠 叶贤涛 顾学范 余永国 肖 冰 孙 昱
上海交通大学医学院附属新华医院 上海市儿科医学研究所内分泌遗传代谢科(上海 200092)
Menke-Hennekam 综合征(Menke-Hennekam syndrome,MKHK)是近年来新发现的常染色体显性遗传病,由CREBBP基因错义杂合变异所致,2016年由Menke等[1]首次报道,迄今为止已报道了约30个病例、20 种CREBBP基因致病变异位点,变异均位于CREBBP基因的30 外显子末端至31 外显子首端(NM_004380.3: c.5128_5614)[1–6]。临床表现为精神运动发育迟缓、不同程度智力障碍、矮身材、小头畸形以及特殊面容(上斜眼睑、内眦距宽、塌鼻梁、短鼻、长人中、招风耳、低耳位、小下颌等)。
Rubinstein-Taybi 综合征(Rubinstein-Taybi syndrome,RSTS)是经典的常染色体显性遗传病,其显著临床特征包括宽指/趾、矮身材,中至重度智力障碍、特殊面容(诡秘微笑,高拱眉、长睫毛、鹰钩鼻、下斜眼睑、高颚等)等[7–8]。RSTS由CREBBP基因的功能丧失(loss of function,LOF)变异(移码变异、无义变异等)所致,变异广泛分布于MKHK 变异区域以外的CREBBP基因区域。
CREBBP不同区域和类型变异导致的RSTS 和MKHK 均有发育迟缓、智力发育落后、小头畸形等临床特征,但特殊面容特征具显著差异。目前对于RSTS 的临床表型、基因型及致病机制均有较多报道,而对于MKHK 的研究较少,并且尚无中国病例的大规模报道。本文总结7 例CREBBP基因变异所致MKHK的临床特征及基因变异特点,进一步扩大MKHK的临床表型谱和基因变异谱。
1 对象与方法
1.1 研究对象
选取2012—2022年就诊于新华医院内分泌/遗传科门诊确诊为Menke-Hennekam 综合征的7 例患儿。诊断符合以下2条:①基因检测,CREBBP基因杂合致病/可能致病变异且位于30外显子末端至31外显子首端区域(NM_004380.3: c.5128_5614);②临床表现,患儿具MKHK 特征性临床表现(特殊面容包括上斜眼睑、内眦距宽、塌鼻梁、短鼻、长人中、招风耳、低耳位、小下颌等)。
本研究获得患儿监护人的知情同意,并经新华医院医学伦理委员会批准(No.XHEC-D-2023-028)。
1.2 方法
1.2.1 临床资料收集 通过回顾门诊、住院及随访资料或联系患儿家属获得临床数据,使用标准化表格收集整理信息,包括一般资料、临床表现、出生与母孕史、家族遗传史、实验室检查结果、影像学检查结果、基因检测结果等。患儿临床表型由具有经验的儿内分泌/遗传科医师进行评估,形态学评估标准参考文献[9–18]。随访资料通过内分泌/遗传科门诊复诊或电话随访采集,随访周期不定,随访内容包括患儿的身高、体重、精神运动发育情况及医疗问题等。
1.2.2 基因检测方法 应用EDTA 抗凝管抽取患儿及家长外周静脉血2 mL 行核心家系外显子组测序(Trio whole exome sequencing,Trio-WES)。采用全基因组DNA 提取试剂盒(Qiagen 公司,德国)提取DNA,用Covaris S220超声破碎仪(Covaries公司,美国)将基因组DNA片段化为200 bp左右片段,用exome research panel(IDT公司,美国)进行捕获,用Illumina NovaSeq 6000测序仪(Illumina公司,美国)对捕获片段进行双端测序(pair-end sequencing),测序片段长度为150 bp。测序产生的fastq数据用BWA比对到hg19参考基因组,按照GATK最佳实践(v3)进行数据分析检测变异。变异用vcfanno 和SNPEff进行注释,排除人群数据库中高频变异,选择错义、无义、剪切位点、框移插入缺失及框内插入缺失作为候选变异。候选变异参考美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南对变异位点进行致病性评估,利用Sanger 测序对家系进行基因变异验证并判断变异的来源。
2 结果
2.1 临床资料
7 例MKHK 患儿4 男3 女;平均就诊年龄(2.6±1.7)岁;平均出生身长(48.8±2.3)cm,平均出生体重(2 548±369)g。除患儿2、4有宫内发育迟缓外,余5 例均无异常出生史,7 例均有产后发育迟滞。
7 例MKHK 患儿均以发育落后就诊,为全面性发育迟缓,以运动及语言发育落后为主。存在多种表观畸形,其中7例存在内眦距宽、招风耳、低耳位,6例存在眼睑上斜、短睑裂、塌鼻、长人中,均无多毛、高鼻梁和诡秘微笑。其余特殊表现见表1。
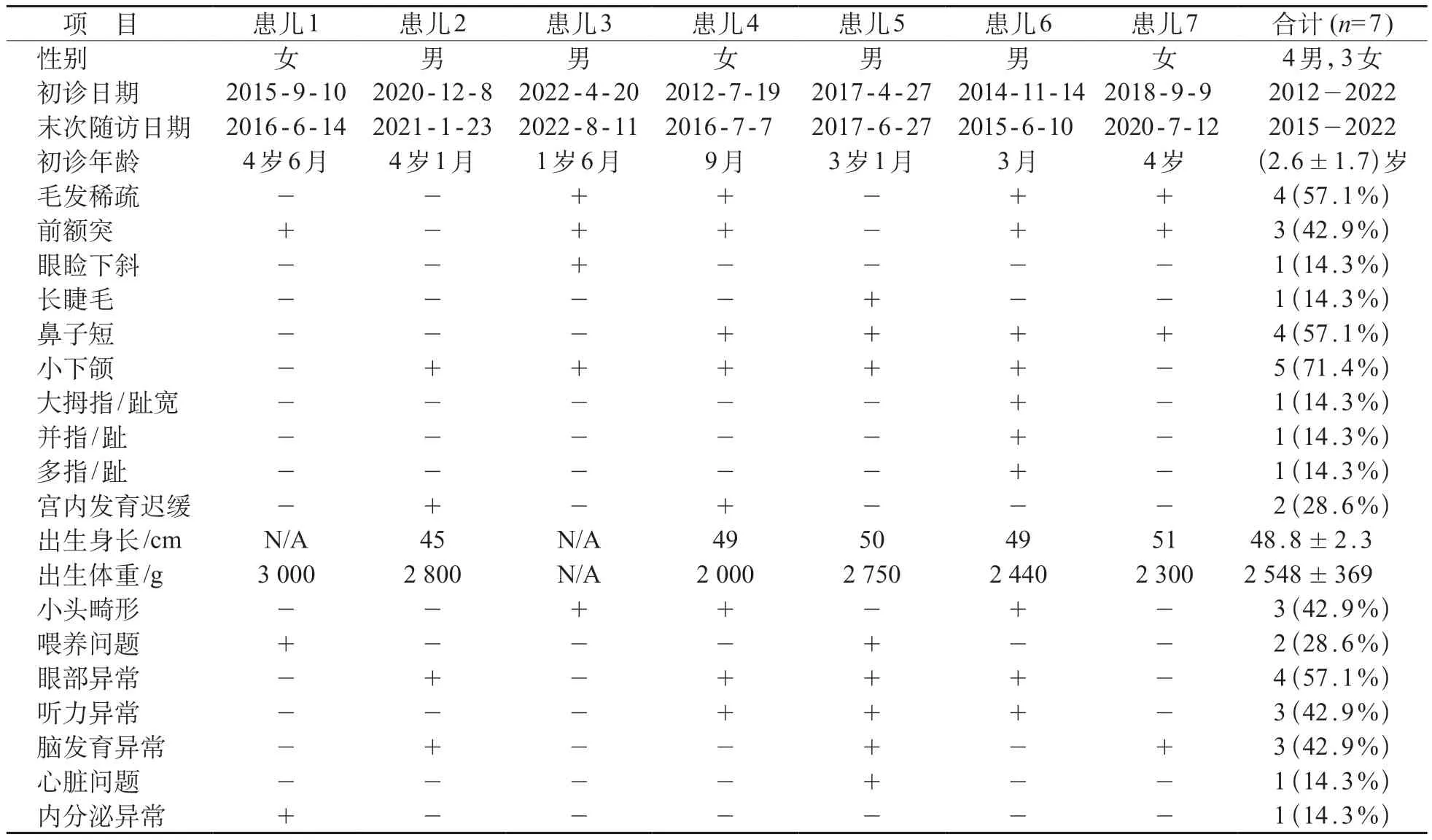
表1 7例Menke-Hennekam综合征患儿临床特征总结
7例患儿随访时长为2个月至4年,平均(11±16)月,末次随访日期见表1。末次随访时身高小于同年龄同性别儿童第三百分位数、体重低于同年龄同性别儿童第三百分位数均为6例(85.7%)。
2.2 基因型分析
7 例患儿的基因检测结果显示均携带CREBBP基因的杂合致病/可能致病变异,共涉及2种类型基因变异(6/7 错义变异、1/7 不影响阅读框的插入缺失),均为新发(de novo),其中2例携带c.5602C>T(NM_004380.3)位点杂合变异。经查询HGMD、ClinVar及PubMed数据库,c.5218C>T与c.5225T>A(NM_004380.3)既往文献未报道过。根据ACMG遗传变异分类标准与指南,对7 例患儿的基因检测结果进行变异致病性分析,共有5 例“致病(P)”、2例“可能致病(LP)”,评析结果见表2。
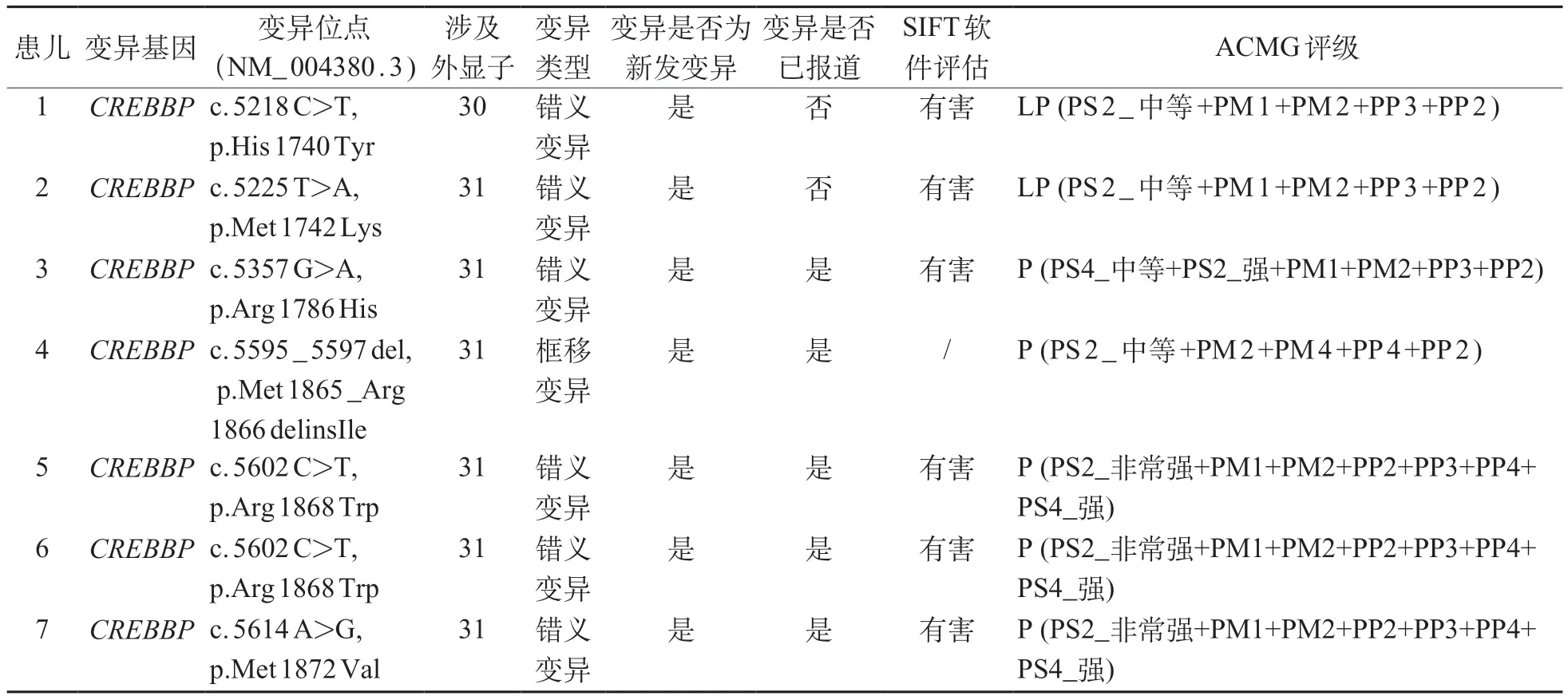
表2 7例Menke-Hennekam综合征患儿基因变异及致病性分析
结合临床表现、家族基因测序及靶向测序验证的结果,7例患儿均诊断为MKHK。
3 讨论
本研究发现2 例CREBBP基因新发错义变异,c.5218 C>T 及c.5225 T>A,为国际上未报道的病例,属于报道较少的MKHK 疾病的变异区域,丰富了CREBBP基因变异谱。MKHK的临床表型异质性强,无统一的临床诊断标准,需结合基因测序结果确定致病原因。目前尚无MKHK中国人群的大型报道,临床医师对此病认识可能不足,容易导致误诊和延迟诊断。
本研究7 例患儿携带的变异均位于CREBBP基因的30外显子末端至31外显子首端区域,且具有不同于RSTS 的特殊面容,本组病例虽然均属亚洲血统,但仍与Menke等[1-2]于2016与2018年报道的疾病表型相近,支持MKHK诊断。既往文献表明,变异位于c.5595_5614(NM_004380.3)区域的MKHK患者具较集中的特殊面容,表现为上睑下垂(33%)、上斜眼睑(58%)、短睑裂(42%)、内眦距宽(54%)、塌鼻梁(54%)、短鼻(50%)、前倾鼻孔(46%)以及长人中(50%),婴儿时期的面容特征最显著,随着年龄增长疾病表型越不明显[2]。本研究患儿4、5、6、7的基因变异位于该区域内,且面容特征与文献报道相符,进一步为该区域变异病例可能属于MKHK独特亚型提供临床依据[2]。另外,相比于其他区域的MKHK 患儿,c.5595_5614 区域变异的患儿具更严重的视听障碍(3/3听力问题、3/3眼部问题),这一发现有待继续收集病例提供证据支持。c.5357G>A是文献报道过的变异,同一碱基位点报道过的变异还有c.5357G>C,相邻区域变异还有c.5354G>A[2,4]。文献指出c.5354 及c.5357 位点变异的MKHK 患者具与RSTS 相似的特征[4],如下斜眼睑、长睫毛、秃鼻梁、宽鼻尖、高颚以及轻度宽指/趾等。本研究c.5357G>A病例的特殊面容为前额突出、面颊饱满、眼睑下斜、眼距宽、宽鼻尖、牙齿不整齐、嘴唇上翻、小下颌,部分表型与上述研究[4]报道相近,需继续收集更多病例以进行表型特征总结。此外本例患儿语言运动发育落后,经康复训练后症状部分改善,也提示早期康复训练对于患者治疗有指导意义。患儿4、6 是本研究中有手指及脚趾畸形者,表现为并指及多趾。2018年Menke等[2]报道中C19病例也有手指皮肤并指表现,但他们都不具RSTS典型的大拇指宽及向桡侧弯曲特征。
目前CREBBP变异导致RSTS的研究较为充分。RSTS 可由CREBBP杂合变异所致,约80%经测序分析确定为基因内的小缺失/插入、错义、无义和剪接位点变异,约20%通过基因靶向缺失/重复分析确定为外显子或整个基因缺失[8,19]。CREBBP基因编码的蛋白是具有乙酰转移酶活性的转录共激活因子,以往研究表明CREBBP缺失可影响转录识别、降低乙酰化活性及破坏染色质重组,继而导致RSTS的发生[20-22]。与MKHK 相关的CREBBP基因变异研究主要与锌指结构域Zinc finger 2(1701-1744)及Zinc finger 3(1765-1846)相关,研究表明这两个介导锌离子结合的结构域都包含半胱氨酸残基,后者可稳定蛋白螺旋折叠并介导与转录因子的结合,c.5170G>A(p.Glu1724Lys)的变异位点与ZNF2的第二个高度保守的半胱氨酸残基(Cys 1723)相连,此变异导致的氨基酸改变引起静电势差,可能对蛋白质相互作用产生重要影响[3],进而解释MKHK的病理机制。
综上,本文报道了CREBBP基因的6个新发变异,其中2 个既往未报道过,进一步扩大了MKHK 的基因变异谱。由于MKHK临床表型异质性强且无国际上认可的诊断指南,诊断较难确立,临床医师在工作中遇到临床表型疑似者,应尽早行高通量测序以协助诊断。
