双核钒配合物催化烯烃聚合的研究
2023-08-14聂金鑫任飞扬陈小建侯小华
聂金鑫, 任飞扬, 陈小建, 侯小华
(合肥工业大学 化学与化工学院,安徽 合肥 230009)
聚烯烃是一类应用十分广泛的高分子材料。近年来高端聚烯烃成为我国聚烯烃产业走向高端化的战略突破点[1]。自1953年齐格勒纳塔催化剂的发现及工业化应用以来[2-3],人们对过渡金属配合物催化烯烃聚合进行了广泛研究,发展出多种能高效催化烯烃聚合的过渡金属配合物催化剂,可应用于合成不同结构的聚烯烃[4]。国内的一些课题组对烯烃配位聚合过程中的聚合机理、立体控制和活性中心结构等方面进行了深入研究,文献[5-11]对催化剂活性中心的形成及失活,聚合物链的引发、增长、终止和转移机理进行了研究。
在常规的聚烯烃分子链中引入极性基团,可以有效地改善聚合物的硬度、黏弹性、界面性质、表面性质、与其他聚合物的共混性能及流变性能等,可使聚合物高附加值化并拓宽其商业用途。目前,功能聚烯烃的合成是聚烯烃领域的重要研究方向[12]。最有效的制备方法是通过非极性烯烃单体与极性单体的共聚反应[13]。目前已知最好的催化剂是含有膦磺酸配体的钯催化剂[14]。但受限于贵金属成本,催化活性以及所得聚合物性能不理想,该类催化体系在实际生产中的应用受到限制。
另一方面,在设计合成具有优异催化性能的催化剂过程中,人们发现双核催化剂体系在烯烃聚合中具有突出表现,包括聚合物分子量、支化程度、共聚单体的插入率以及立体选择和化学选择性等方面[15]。一般来说,双核催化剂都表现出比单核类似物更高的催化活性,得到分子量更高的聚合物,更高的共聚单体插入率。这种催化结果归因于金属活性中心之间的协同效应以及聚合物链与相邻的金属中心发生的次级作用。虽然很多团队通过理论计算对聚合过程进行推测,但其结论仅适用于当下特定的双核配合物结构,对其金属活性中心之间的相互作用仍缺乏认识。
目前报道的双核催化剂在结构设计上涉及多种过渡金属,代表性的催化剂结构如图1所示。但是对于双核钒配合物催化剂的研究报道仍然很少。自1958年文献[16]报道了VCl4-AlBr3-AlPh3体系能够高效催化乙烯聚合以来,此后研究者们发展出一系列高效钒配合物催化剂[17],尤其是日本Nomura课题组报道的一系列五价钒配合物能够高效催化多种烯烃发生聚合[18]。基于双核配合物和单核钒催化剂在功能化聚烯烃合成中的潜在优势,本文设计合成了一系列具有不同结构的链接基团的双核钒配合物,并研究其在烯烃聚合中的催化性能。

图1 代表性双核配合物烯烃聚合催化剂
1 实验部分
1.1 主要原料
2,6-二甲基苯基异氰酸酯、三氯氧钒、4,4′-联苯二醇、五氟苯酚等药品按纯度需求均购于阿拉丁试剂(上海)有限公司,使用前未经纯化处理;6-氯-1-己烯、10-十一烯酸甲酯(UA)、10-十一烯醇(UOH) 均购于西格玛公司,使用前经干燥减压蒸馏纯化;商用乙烯购于南京上元气体有限公司,直接用于聚合反应;所用甲苯、正己烷、正辛烷溶剂在钠丝和二苯甲酮存在下进行回流12 h至紫黑色后蒸馏收集,随后使用0.4 nm分子筛浸泡过夜后待用。
1.2 实验仪器及测试条件
实验所有对空气和水敏感的实验操作,均在干燥的氮气气氛下进行标准Schlenk操作或在手套箱中完成;配合物的核磁共振谱(nuclear magnetic resonance,NMR)包括1H NMR、13C NMR和19F NMR,通过德国布鲁克公司生产核磁共振仪进行表征获得;聚合物分子量、分子量分布数据通过凝胶渗透色谱(gel permeation chromatography,GPC) 分析,由美国安捷伦公司的1260lnfinity Ⅱ HT GPC获得;检测条件:流动相为1,2,4-三氯苯(色谱级),温度150 ℃,流速0.3 mL/min,Mn和聚合物分散性指数(polymer dispersity index,PDI)数据参照聚苯乙烯标准物获得。傅里叶变换红外光谱(Fourier transform infrared,FTIR)数据通过美国Thermo Nicolet公司的Nicolet 67型傅里叶红外光谱仪表征获得;聚合物熔点通过差示扫描量热法(differential scanning calorimetry,DSC)表征,使用差示扫描量热仪 Q2000 (TA Instruments);聚合物样品于氮气氛围(50 mL/min)的环境下,测试温度范围为-50~200 ℃,升温速率为10 ℃/min,降温速率同样为10 ℃/min,循环测试3次;聚合物Tm取第2次测得的数值;元素分析是通过埃克塞特分析公司的EAICE-440 C/H/N/O/S元素分析仪进行表征测试。
1.3 实验方法
1.3.1 双核钒配合物B1的合成
称取双核钒三氯化物 (426 mg,0.99 mmol) 溶于甲苯 (15 mL) 中,-30 ℃ 静置1 h后,加入五氟苯酚锂盐(131.5 mg,0.54 mmol),溶液颜色由墨绿色转为深褐色,溶液温度逐渐上升到室温后继续搅拌12 h后停止,将溶液经过装有硅藻土的砂芯漏斗过滤,滤饼用少量甲苯洗涤直至流出液无色,合并所得滤液并抽干。将粗品溶于甲苯和己烷混合溶剂,-30 ℃下重结晶得到红褐色固体(625 mg,产率80%)。1H NMR (CDCl3,25 ℃):δ7.49×10-6(d,J=6.0 Hz, 2H, Ar—H),7.18×10-6(d,J=12.0 Hz, 2H, Ar—H),7.08×10-6(d,J=6.0 Hz,2H,Ar—H),7.03×10-6(d,J=6.0 Hz,2H,Ar—H),4.08×10-6(br,2H,—CH2);13C NMR (CDCl3,25 ℃):δ166.6×10-6,163.3×10-6,161.8×10-6,160.9×10-6,158.8×10-6,138.7×10-6,136.8×10-6,119.2×10-6,113.4×10-6,111.3×10-6,104.2×10-6,43.3×10-6;19F NMR (CDCl3,25 ℃):δ-136×10-6,-153×10-6,-162×10-6。对C25H10Cl4F10N2O2V2进行元素分析,各元素质量分数计算值分别为:C 37.35%,H 1.25%,N 3.48%; 实验值分别为:C 37.66%, H 1.45%, N 3.23%。
1.3.2 双核钒配合物B2的合成
称取单核钒三氯化物 (150 mg,0.54 mmol)溶于甲苯 (15 mL) 中,-30 ℃ 静置1 h后,分批加入4,4′-联苯二酚锂盐 (53.5 mg,0.27 mmol),溶液颜色由墨绿色转为深褐色,室温搅拌12 h后停止反应。用装有硅藻土的砂芯漏斗过滤得到深褐色溶液,减压移除溶剂后的粗产物在己烷和甲苯溶解,-30 ℃下重结晶获得红褐色固体产物(126 mg,产率70%)。1H NMR (CDCl3,25 ℃):δ7.48×10-6(d,J=12.0 Hz,4H, Ar—H), 7.14×10-6(d,J=12.0 Hz,4H,Ar—H),6.91×10-6(br,6H,Ar—H),2.57×10-6(br,12H,—CH3);13C NMR (CDCl3,25 ℃):δ162.7×10-6,152.5×10-6,150.4×10-6,139.6×10-6,129.7×10-6,127.5×10-6,123.1×10-6,116.9×10-6,18.6×10-6。对C28H26Cl4N2O2V2进行元素分析,各元素质量分数计算值分别为:C 50.48%, H 3.93%, N 4.20%;实验值分别为:C 50.68%, H 4.16%, N 4.06%。
1.3.3 双核钒配合物B3的合成
B3的合成通过与B2相同的步骤进行,不过将4,4′-联苯二酚锂盐换成亚甲基双酚锂盐(92.1 mg,0.27 mmol),得到红褐色固体 (193 mg,产率88%)。1H NMR (CDCl3, 25 ℃):δ6.88×10-6(m,3H,Ar—H),2.87(s,6H,—CH3);13C NMR(CDCl3,25 ℃):δ165.8×10-6,164.5×10-6,162.1×10-6,161.8×10-6,157.6×10-6,137.4×10-6,128.4×10-6,127.5×10-6,127.3×10-6,126.6×10-6,19.3×10-6;19F NMR(CDCl3,25 ℃):δ-139×10-6,-156×10-6,-160×10-6。对C28H18Cl4F8N2O2V2进行元素分析,各元素质量分数计算值分别为:C 41.51%, H 2.24%, N 3.46%;各元素质量分数实验值分别为:C 41.77%, H 2.39%, N 3.25%。
1.4 聚合过程
降冰片烯聚合反应过程如下:称量降冰片烯(200 mg,2.12 mmol)于15 mL螺口瓶中,加入4.5 mL甲苯将其溶解;依次加入包含B1 (0.30 mL,0.5 μmol)的甲苯溶液和助催化剂AlMe3溶液(2.0 mL,2.0 mmol);然后搅拌溶液至规定的时间,添加过量的苯甲醛(PhCHO)终止聚合反应,搅拌30 min后将混合溶液倒入含有稀盐酸的100 mL甲醇中沉出聚合物;过滤收集白色固体并用甲醇洗涤3次,室温下进行真空干燥后称重。
乙烯均聚和共聚反应过程如下:从高温烘箱中取出100 mL规格高压反应釜釜体,搭接好高压反应釜装置,氮气置换3次;油浴锅加热聚合釜到设定温度后,氮气保护下用注射器依次将助催化剂溶液和催化剂溶液注入反应釜中;关闭进样阀门,打开乙烯阀门通入乙烯开始聚合反应,调节并保持反应釜体系内的压力为4个大气压,剧烈搅拌反应溶液至设定的反应时间;关闭乙烯气体阀门,释放反应釜中乙烯气体后打开反应釜,将聚合溶液倒入约100 mL含有稀盐酸的甲醇中;搅拌2 h后,过滤收集所得固体并用甲醇洗涤3次;所得聚合物于60 ℃下真空干燥6 h后称重。
2 结果与讨论
2.1 双核钒配合物的合成
本文设计合成一系列骨架可调的双核钒二氯合物。通过采用不同的骨架实现调节金属原子之间距离,实现对金属原子周围的位阻和电子环境的调节,为研究双金属中心之间的相互作用提供可能。合成过程如图2所示。

图2 双核钒二氯化物的2种设计合成路线
从图2a可以看出,酰亚胺配体能够稳定高氧化态的钒配合物,通过二异腈酸酯与过量的三氯氧钒发生反应,制备相应双核钒三氯化物,再与五氟苯酚锂盐反应,生成目标双核钒二氯化物B1。从图2b可以看出,使用双酚试剂的相应锂盐为反应物,与已制备的酰亚胺钒三氯化物按照精确投料比1∶2进行反应,得到双核钒的二氯化物(B2、B3)。
2.2 双核钒二氯化物催化乙烯聚合
以双核钒二氯化物为前体,在烷基铝助催化剂的活化下可生成金属阳离子活性中心,从而催化乙烯发生聚合。本文研究助催化剂种类和用量、聚合反应温度、催化剂结构对催化活性及聚合产物的影响,聚合结果见表1所列。表1中聚合实施的一般条件如下:双核钒催化剂用量为0.5 μmol,单核钒配合物的用量为2.0 μmol,助催化剂为氯化二乙基铝溶液(Et2AlCl),溶剂为甲苯且总体积为10 mL,乙烯压力为4×105Pa,反应时间为5 min,c(Al)/c(V)为聚合溶液中Et2AlCl与催化剂的浓度之比,实验组6的聚合条件需额外加入0.2 mmol三氯乙酸乙酯。 对于所得聚合物分子量的表征,是将高温溶解在1,2,4-三氯苯的聚合物溶液过滤后,通过高温GPC以苯乙烯为参照样品测得。
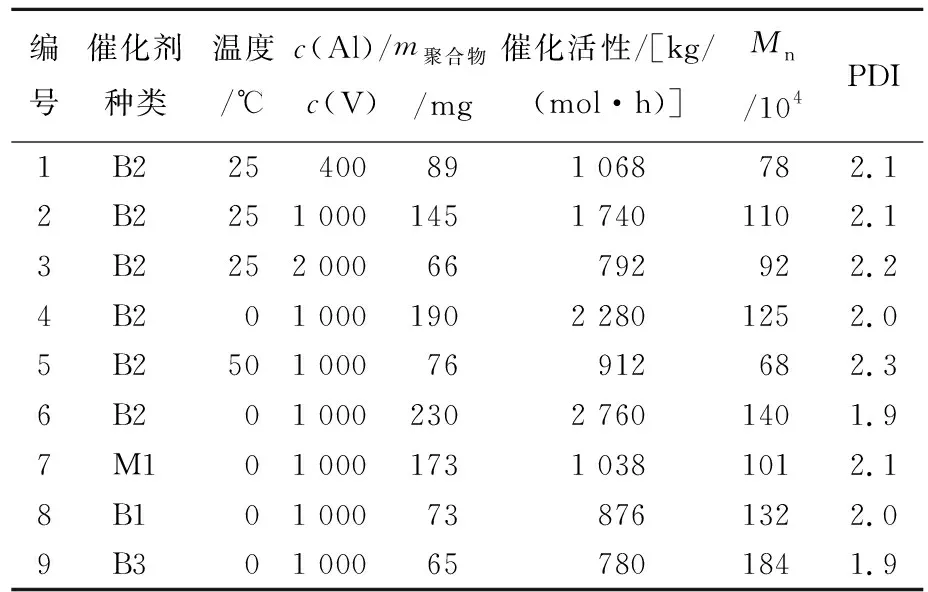
表1 双核钒二氯化物催化乙烯聚合结果
聚合结果表明,不同的助催化剂具备明显不同的活化能力,加入AlMe3、AlEt3或AliBu3均得到少量聚合物。而Et2AlCl的使用可有效实现钒氯化物的活化,得到大量的聚合物。助催化剂的投量也对聚合活性有很大影响,除了能够与金属配合物反应生成真正的阳离子活性中心,同时还能够进一步除去反应体系中残留的水和氧,从而保证聚合过程顺利发生。经过筛选最佳的用量比例c(Al)/c(V)=1 000。不同温度下的聚合结果表明,在0 ℃下催化剂具有最高的催化活性,当聚合温度提高后催化活性明显降低。其原因可能是钒配合物相应的活性中心在高温时不稳定。另外,五价钒配合物催化剂在烯烃聚合反应中往往会被还原成低价态,从而可能导致其催化能力降低。因此可以加入适量的三氯乙酸乙酯有助于提高催化活性[13]。
为了探究单核和双核催化剂的催化能力,本文开展了具有类似结构的单核配合物V(N-2,6-Me2C6H3)Cl2(O-2,6-Me2C6H3)(M1)的乙烯聚合实验。相比单核催化剂,双核催化剂B2具有更高的催化活性。可能原因是联苯基团的引入使得钒金属活性中心的电子云密度降低,提高了其与乙烯单体的反应能力。
对于不同结构的双核催化剂,B1和B3表现更低的催化活性,可能原因是强吸电子基团的引入会导致金属阳离子活性中心不稳定,但其与烯烃单体的反应能力提高,得到聚合物分子量更高。以上结果表明,双核钒配合物在超高分子量聚乙烯合成制备中具有潜在的应用价值。
2.3 双核钒二氯化物催化降冰片烯聚合
在降冰片烯聚合反应中,本文使用不同类型的烷基铝助催化剂,发现用AlMe3作为助催化剂时,聚合溶液黏度明显增大,加入甲醇后沉出大量聚合物。相同反应时间下,使用助催化剂AlEt3、Et2AlCl或AliBu3,均未获得明显聚合物。尤其是降冰片烯聚合需要在较高的温度下进行。不同温度的聚合结果见表2所列。
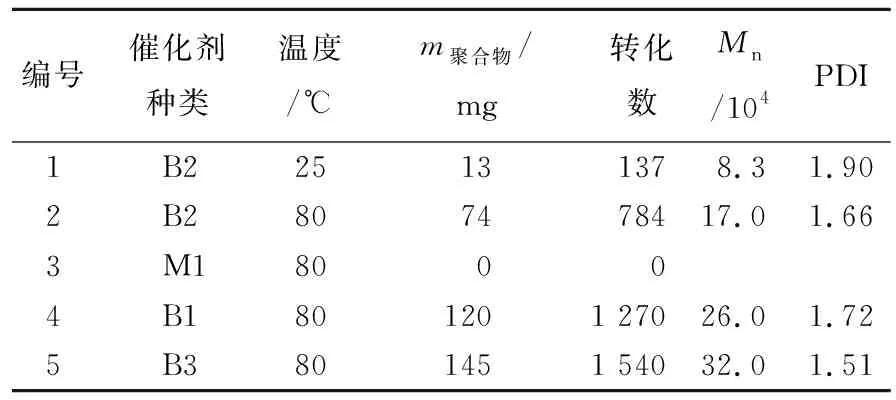
表2 双核钒二氯化物催化降冰片烯聚合结果
表2的聚合条件如下:聚合时间为1.5 h,降冰片烯用量为2.12 mmol, 双核钒配合物物质的量为0.5 μmol,甲苯总体积为10 mL,使用AlMe3为助催化剂,c(Al)/c(V)=1 000,常温GPC测得聚合物分子量,以苯乙烯为参照样品。该聚合产物能在四氢呋喃中具有良好的溶解性,而且GPC结果所得聚合物分子量分布较窄(PDI为1.50~1.70)。
为探究降冰片烯发生聚合方式,可以通过核磁共振表征确定聚合物结构,如图3所示,1H NMR条件为600 MHz,试剂CDCl3,25 ℃。图3中,*表示残留少量杂质峰。由图3可知,聚降冰片烯为开环易位聚合的结构,没有发现配位插入的结构单元。
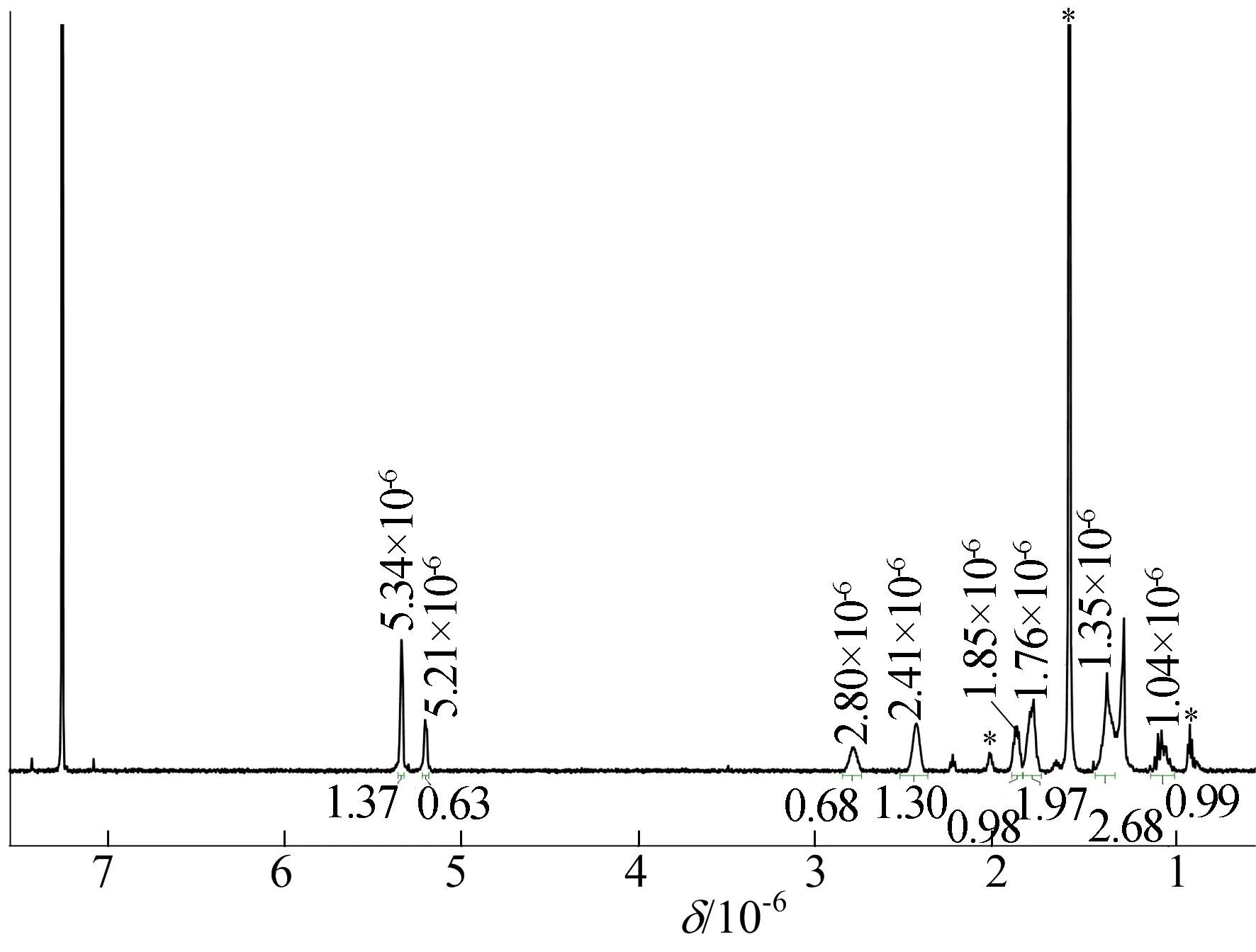
图3 聚降冰片烯的1H NMR谱图
根据相关报道,钒的氯化物与三甲基铝反应生成二甲基化产物,随后高温下可能发生α-H消除,生成钒卡宾配合物[19],从而引发降冰片烯发生开环易位聚合,反应过程如图4所示。同样的反应条件下,单核催化剂M1未能得到聚合产物。而对于B1和B3配合物,可能由于其含有强的吸电子基团(—OC6F5)导致其更易形成钒卡宾结构,从而表现出更高的聚合物产率。
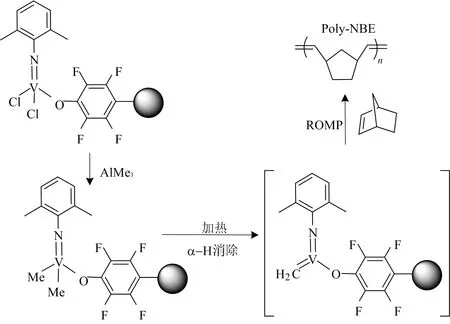
图4 钒配合物催化降冰片烯发生开环易位聚合过程
2.4 双核钒二氯化物的催化能力
为进一步研究双核钒配合物在乙烯和极性单体共聚反应中的催化能力,本文选取具有代表性的3种极性单体(6-氯-1-己烯、10-十一烯醇、10-十一烯酸甲酯)用于实验,反应过程如图5所示。
双核钒二氯化物催化乙烯与极性单体共聚结果见表3所列。表3中双核钒催化剂B2的用量为2.0 μmol,所用助催化剂为Et2AlCl,c(Al)/c(V)=1 000,共聚单体投量为1.5 mmol,甲苯总体积为10 mL,乙烯压力为4×105Pa,反应时间为5 min,反应温度为0 ℃,实验组1中双核钒催化剂用量为0.5 μmol。由表3可知,相比于乙烯均聚反应,双核钒催化剂在加入1.5 mmol的6-氯-1-己烯(Cl-Hex)的共聚反应中的活性没有明显变化,说明氯原子并未影响聚合反应过程。

表3 双核钒二氯化物催化乙烯与极性单体共聚
在乙烯与极性单体十一烯醇(UOH)和十一烯酸甲酯(UA)的共聚反应中,双核钒配合物的催化活性明显降低。这可归结于极性基团与金属中心的螯合作用,抑制了碳碳双键的配位插入[20]。过量助催化剂可以起到掩蔽剂的作用,能将极性单体的羟基或酯基进行保护,减弱了其对金属活性中心的毒化作用。聚乙烯和共聚物的DSC表征结果如图6所示。
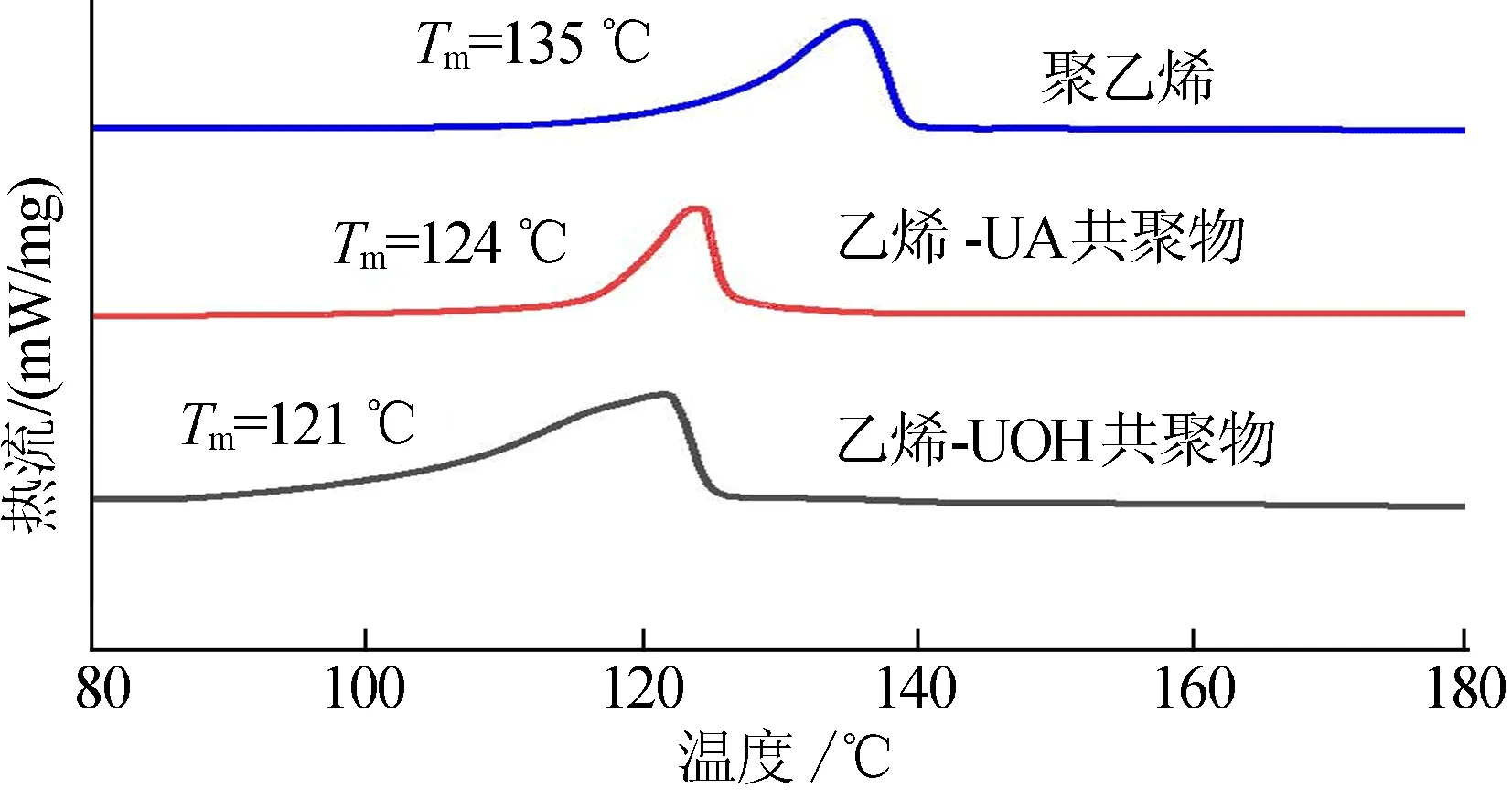
图6 聚乙烯与共聚物的DSC曲线
相比于聚乙烯,共聚物的熔融转变温度明显下降,结果证明共聚单体的成功插入。相应的聚合物1H NMR谱图(500 MHz、C2D2Cl4、120 ℃)如图7所示。
由图7可知,通过特征峰的积分计算共聚单体的插入率,值得注意的是,双核钒配合所制备的共聚物具有很高的分子量[20]。
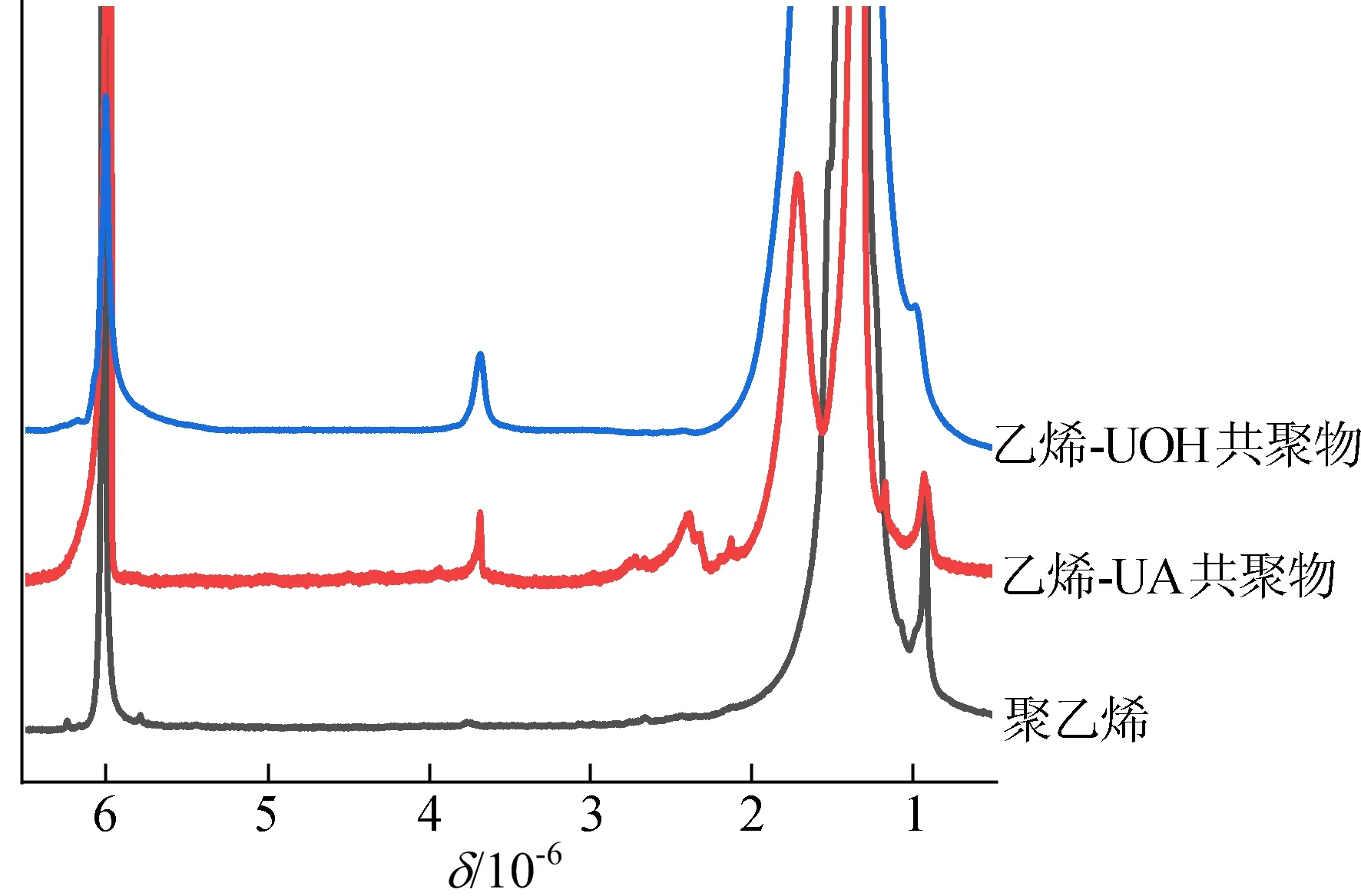
图7 共聚物与聚乙烯的高温1H NMR谱图
3 结 论
本文制备了3种双核钒的二氯化物并进行表征。在三甲基铝为助催化剂时,双核钒配合物催化降冰片烯发生开环易位聚合,含有五氟苯酚基团的配合物表现出更高的催化活性;而在乙烯聚合中需要使用氯化二乙基铝为助催化剂,双核比单核配合物具有更高的催化活性,并得到超高分子量聚乙烯;在乙烯与十一烯醇或十一烯酸甲酯的共聚反应中,双核配合物B2中仍表现出中等催化活性,制得高分子量的共聚物且共聚单体的插入率为0.6%~1.2%。
