城乡交错区的土壤微生物多样性格局及其影响因素
2023-05-30高燕,王嵘
高 燕, 王 嵘
(华东师范大学 浙江天童森林生态系统国家野外科学观测研究站, 浙江 宁波 315114)
0 引 言
生物多样性对人类可持续发展至关重要, 供给人类所需的各种生态系统服务[1-2]. 微生物是地球生物多样性最丰富的资源, 广泛存在于各类基质中, 如空气、水和土壤等. 其中, 土壤微生物在碳、氮等元素的生物地球化学循环中发挥着关键作用[3-4]. 其通过不同途径改变着土壤的物理、化学和生物学特性, 包括有机质的分解与合成[5]、养分的释放与固定[6]、污染物的降解[7]等, 主导和参与陆地生态系统的一系列重要生态过程[8-9]. 土壤微生物多样性是维持生态系统功能, 提供生态系统服务的重要环节,其多样性丧失将对陆地生态系统的气候调节[10]、养分循环及粮食生产[8]等服务功能产生负面影响[11-12].因此, 明确土壤微生物多样性格局及其驱动因素对维持生态系统的稳定性具有重要指示意义.
改革开放以来, 我国处在快速城市化发展阶段. 据人口统计数据显示, 至2018 年我国城市人口占总人口的比例为59.6%, 伴随城市区域的不断扩张, 我国绝大多数人口将居住在城市中[13-14]. 城乡交错区是城市边缘区域, 是城市生态系统向农村生态系统转变的过渡带, 兼具两者的景观结构[15], 既有大量的居民居住区和公园等城市景观, 也有较大面积的农业用地[16]. 城乡交错区独特的地理位置, 使该区域受城市扩张影响剧烈, 土地利用格局高度复杂, 呈现出景观异质性和破碎化程度均高于城市和农村生态系统[17-18]. 因此, 研究城乡交错区生态系统中生物多样性的分布格局与维持机制对城市生态文明建设与人类福祉至关重要[19].
城乡交错区的重要服务功能有为中心城区供应蔬菜、粮食等食物资源, 作为中心城区的生态屏障等[20]. 土壤微生物多样性是植物多样性和生产力的重要驱动因素, 土壤微生物会通过营养物质的矿化和竞争等作用影响植物的营养供应和资源分配[21], 从而维持城乡交错区的生态系统服务[22]. 因此, 全面地对土壤微生物类群开展研究可以掌握土壤微生物多样性的整体格局, 为城乡交错区的生物多样性保护提供针对性建议.
土壤微生物多样性的分布格局在大尺度上的分布规律已得到深入研究. 全球尺度上, 土壤真菌多样性符合纬度地带性分布, 土壤细菌多样性在中纬度地区达到最高值, 在极地和赤道地区呈下降趋势[23];大陆尺度上, 亚洲森林的土壤细菌多样性分布格局与土壤酸碱度显著相关[24]; 局域尺度上, 我国华北平原的土壤微生物多样性分布格局主要受到地理距离的调控[25]. 城市生态系统中, 土壤微生物丰富度的分布格局与城市化程度呈正相关[26], 然而在城乡交错区这样的小尺度范围内, 土壤微生物多样性的分布格局尚不清晰.
为全面揭示城乡交错区土壤微生物的多样性分布格局及其影响因素, 本文以上海一城乡交错区为研究对象, 揭示城乡交错区常见土地利用类型中主要土壤微生物类群(土壤细菌和土壤真菌)的多样性分布格局与潜在驱动因素.
1 材料与方法
1.1 研究样地与土样采集
研究地点位于上海市松江区泖港镇(121°12′ E ~ 121°28′ E, 30°94′ N ~ 30°96′ N), 属于亚热带季风气候, 四季分明, 雨量充沛, 年平均气温15.4℃, 年均降水量1103.2 mm, 土壤类型为水系不同沉积母质发育的水稻土和灰潮土. 上海市是我国城市化水平最高的城市, 城市化率已达到80%, 具有高不透水面积比例和高土地利用率的特点[27]. 松江区泖港镇位于上海市的中心城区以外, 兼具居民居住区、坑塘水域等城市土地利用类型和耕地、仓储用地等农村土地利用类型, 土地利用现状呈现典型的城乡交错区复杂的土地利用格局. 当地主要植物种类包括杠板归(Polygonum perfoliatum)、黄鹌菜(Youngia japonica)、葎草(Humulus scandens)、南苜蓿(Medicago polymorpha)、苦苣(Sonchus oleraceus)、蛇床(Cnidium monnieri)、菵草(Beckmannia syzigachne)、羊蹄酸模(Rumex japonicus)、野豌豆(Vicia sepium)和芦苇(Phragmites australis), 均为本地常见草本植物.
研究选择泖港镇的耕地(Farm)、坑塘水域(Fish pond)、仓储用地(Granary)和居民居住区(Residence) 这4 种常见的土地利用类型, 其中, 仓储用地的土地使用时长超过20 年. 分别设置5、6、6 和6 个采样点作为统计学重复, 共采集23 份土壤样品. 土壤样品的采集选择对角线法, 在去除地表凋落物后, 使用环刀法采集20 cm 浅层土壤, 混合每个采样点采集到的土壤样品作为1 份混合土壤样品. 土壤样品用干冰保存运回实验室, 在每份土壤样品中选取部分土壤用于理化指标的测定, 其余土壤置于超低温冰箱(—80℃)保存, 用于分析土壤细菌和土壤真菌群落.
1.2 土壤理化指标测定
1.2.1 土壤酸碱度测定
土壤酸碱度(土壤pH)选用电极法测定[28-29], 土壤悬浊液的水土比为2.5∶1, 采用便携式pH 计测定土壤酸碱度值.
1.2.2 土壤孔隙度测定
首先测定土壤容重和土壤比重, 然后根据上述两项的数值计算土壤孔隙度[28-29]. 土壤容重采用环刀法测定[28-29]. 将装有土壤样品的环刀去除顶盖, 放入烘箱, 在(105±2)℃下烘干至恒重, 并称重. 土壤容重的计算公式为
式(1)中:dv为土壤容重(g/cm3),W为烘干后环刀重 + 干土重(g),W环为环刀重(g),V为环刀的体积(cm3).
土壤比重采用比重瓶法测定[28-29]. 将蒸馏水煮沸5 min, 除去水中的CO2, 冷却至室温后, 移入比重瓶中, 并用温度计测定瓶内水温. 然后倒出约一半的水量, 将10 g 过筛烘干土样经干漏斗倒入比重瓶,样品与水均匀混合后加热比重瓶, 沸腾后保持加热1 h. 加热完成后取下比重瓶, 待比重瓶内液体澄清后, 称重并测定瓶内水温. 土壤比重的计算公式为
式(2)中:ds为土壤比重(g/cm3),dt为t℃时蒸馏水比重(g/cm3),M为烘干样品重(g);M1为t℃时比重瓶重 + 水重(g).
最后, 通过式(3)计算土壤孔隙度,
式(3)中:Pt为土壤总孔隙度.
1.2.3 土壤含水率测定
土壤含水率采用烘干法测定[28-29]. 称取土壤鲜样10 g, 置于已知重量的铝盒中, 放入烘箱, 在105 ~110℃下烘干至恒重, 取出后放入干燥器冷却. 冷却后取出铝盒, 盖好盒盖, 称重. 土壤含水率的计算公式为
式 (4) 中:W'为含水率,m0为铝盒重(g),m1为铝盒重 + 湿样重(g),m2为铝盒重 + 烘干样品重(g).
1.3 环境微生物多样性
1.3.1 环境微生物DNA 提取
土 壤 基 因 组DNA 使 用FastDNARSPIN Kit for Soil (MpBiochemial, USA)试 剂 盒 提 取, 用NanoDrop2000 检测土壤基因组DNA 纯度和浓度. 1%琼脂糖凝胶电泳检测土壤基因组DNA 完整性.
1.3.2 环境微生物多样性检测
通过检测16S rRNA 和ITS基因的序列变异及丰度信息, 明确土壤样本中土壤细菌和土壤真菌的多样性信息. 以纯化后的DNA 为模板分别对土壤细菌16S rRNA 基因的V3、V4 区域和土壤真菌ITS基因的ITS1 区域进行扩增. 16S rRNA 基因的扩增引物序列为338F (5’-ACTCCTACGGGAGG CAGCAG-3’), 806R (5’-GGACTACHVGGGTWTCTAAT-3’).ITS基 因 的 扩 增 引 物 序 列 为ITS1F(5’-CTTGGTCATTTAGAGGAAGTAA-3’), ITS2R (5’-GCTGCGTTCTTCATCGATGC-3’). PCR 反应条件: 95℃预变性3 min; 29 ~ 35 个循环; 循环包括95℃变性30 s, 53 ~ 55℃复性30 s, 72℃延伸45 s; 最后72℃延伸5 min. 使用AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City,CA, USA)纯化回收产物, 用2%琼脂糖凝胶电泳检测PCR 产物, 并用Quantus Fluorometer(Promega, USA)检测定量回收产物. 使用NEXTFLEX Rapid DNA-Seq Kit 构建测序文库, 采用MiSeq PE300 高通量测序平台(Illumina, USA)进行测序.
1.4 数据分析
1.4.1 土壤理化指标分析
首先检验所有土壤理化指标的多重共线性, 采用方差膨胀因子分析(variance inflation factor,VIF, 表示为V) 对某一因子和其余因子进行回归, 得到R2值, 并使用R version 4.1.0 分析软件car version 3.0-11 包中的vif 函数计算V值[30].V值越大, 相关性越强, 当V> 8 时判定该因子共线性强,需剔除该因子, 最终保留低相关性的因子以增强整体解释能力. 继续计算并检验土壤理化指标在各土地利用类型间的差异情况, 采用的分析方法是最小显著差数(least significant difference, LSD)检验法,调用R version 4.1.0 分析软件中的agricolae1.3-5 完成上述分析[31].
1.4.2 高通量测序数据质控与土壤微生物鉴定
使用软件Trimmomatic version 0.40[32]对原始数据进行质控, 过滤reads 尾部质量值20 以下的碱基, 设置窗口为50 bp, 若窗口内的平均质量值低于20, 即从窗口开始截去后端碱基, 过滤质控后50 bp以下的reads, 并去除含N 碱基的reads. 采用软件FLASH version 1.2.11 (https://ccb.jhu.edu/software/FLASH/index.shtml)完成拼接, 设置最小overlap 长度为10 bp, 根据PE reads 之间的overlap 关系将成对reads 拼接成一条序列. 拼接序列的overlap 区允许的最大错配率为0.2, 筛除不符合的序列. 根据序列首尾两端的barcode 和引物区分样品, 调整序列方向, barcode 允许的错配数为0, 最大引物错配数为2, 获取有效数据.
使用软件UPARSE version7.1 (http://drive5.com/uparse), 以97%相似性序列聚为OTUs 的原则对OTUs 进行聚类并剔除嵌合体. 通过软件RDP classifier version 2.11 (https://sourceforge.net/projects/rdp-classifier)对每条序列进行物种分类注释, 设置比对阈值为70%, 在各分类学水平上统计每个样品的群落组成信息, 完成物种注释分析.
1.4.3 土壤微生物的群落组成分析
分别在门、纲、目、科和属的分类学水平上, 剔除已完成物种注释的微生物未命名(norank,AKYG587, JG30-KF-CM45)或没有分类信息(unclassified)的微生物, 仅保留分类信息完整且科学命名的微生物. 检验不同土地利用类型间土壤细菌和土壤真菌OTUs 数目的差异选择Wilcoxon 秩和检验法, 并采用错误发现率(false discovery rate)校正检验结果, 用Q值表示组间差异的显著度. 该分析使用R version 4.1.0 分析软件中的wilcox.test 和p.adjust 函数完成. 通过Shannon 多样性指数绘制稀释曲线, 以该指数伴随着随机抽取序列数增加而变化的情况判断测序数据量是否充分. 采用软件USEARCH version 7.0 (http://www.drive5.com/usearch)中的alpha_div_rare 命令计算得到数值, 并通过R version 4.1.0 分析软件的amplicon version 1.11.1 分析包中alpha_rare_curve 函数绘制稀释曲线[33]. 土壤细菌和土壤真菌的群落组成根据筛选后的物种注释信息选择门和属两个分类学水平上的微生物信息, 使用R version 4.1.0 分析软件中的amplicon version 1.11.1 分析包绘制堆叠柱状图[33], 反映不同土地利用类型中土壤细菌和土壤真菌的群落组成信息和各微生物类群的相对多度情况.
1.4.4 土壤微生物α 多样性分析
选择Chao1、Shannon 和Coverage 指数表征土壤细菌和土壤真菌的α 多样性. 调用R version 4.1.0 分 析 软 件vegan version 2.5-7 包 计 算 各 多 样 性 指 数 并 使 用amplicon version 1.11.1 包 的alpha_boxplot 函数绘制箱型图[33-34]. 土壤细菌和土壤真菌α 多样性指数的土地利用类型间差异选择Wilcoxon 秩和检验法, 计算错误发现率以校正检验结果, 组间差异的显著度用Q值表示. 采用R version 4.1.0 分析软件中的wilcox.test 和p.adjust 函数完成Wilcoxon 秩和检验与错误发现率分析.
1.4.5 土壤微生物β 多样性分析
使用Bary-curtis 距离表征β 多样性, 使用R version 4.1.0 分析软件中vegan2.5-7 包的vegdist 函数计算该指数的距离矩阵, 调用ape5.5 包中的pcoa 函数完成主坐标分析, 并用ggplot 包完成绘图, 呈现土壤细菌和土壤真菌的β 多样性分布格局[34-35]. 选择置换多因素方差分析(permutational multivariate analysis of variance, PERMANOVA)确定对土壤细菌和土壤真菌β 多样性分布格局解释度最高的变量(土地利用类型与土壤理化指标), 使用R version 4.1.0 分析软件中vegan version 2.5-7包的pairwise.adonis 函数计算各变量的解释度[34]. 为了进一步明确对各分组间差异贡献最大的微生物, 选择多级物种判别分析(linear discriminant analysis effect size, LEfSe), 通过Kruskal-Wallis 秩和检验定位到不同土地利用类型间丰度差异显著的微生物, 再利用线性判别分析(linear discriminant analysis, LDA, 表示为A)评估土地利用类型间显著差异的微生物对土地利用类型间差异的贡献度.该分析使用的是Python 中的LEfSe 包, 选择门和属两个分类学水平, 与土壤细菌和土壤真菌的群落组成分析相对应[36]. 此外, 采用vegan2.5-7 包的anosim 函数检验不同土地利用类型间土壤细菌和土壤真菌群落组成的差异[34].
1.4.6 土壤理化环境对土壤微生物多样性和群落组成影响的分析
使用回归分析探究影响土壤细菌和土壤真菌α 多样性分布格局的驱动因素, 调用lm 函数检验土壤细菌和土壤真菌的α 多样性与上述各土壤理化环境指标之间的相关性. 土壤细菌和土壤真菌β 多样性分布格局的驱动因素分析选择冗余分析(redundancy analysis, RDA)或典范对应分析(canonical correlation analysis, CCA). 模 型 的 选 择 参 照 去 趋 势 对 应 分 析(detrended correspondence analysis,DCA)的结果, 如果lengths of gradient 的第一轴大于等于3.5 选择典范对应分析, 否则选择冗余分析.使用vegan version 2.5-7 包的decorana 函数选择模型, rda 和cca 函数分别实现冗余分析和典范对应分析[33]. 调用cor 函数分别计算优势土壤细菌和优势土壤真菌与土壤理化环境指标之间的斯皮尔曼相关系数, 使用pheatmap1.0.12 包的pheatmap 函数绘制热图[37]. 上述分析均使用R version 4.1.0 分析软件完成.
2 结 果
2.1 不同土地利用类型的土壤理化性质
方差膨胀因子分析结果显示, 本研究的土壤理化指标土壤酸碱度(V= 1.12)、土壤孔隙度(V=2.78)和土壤含水率(V= 2.78)之间不存在多重共线性, 3 种土壤理化指标都应保留以继续分析. 研究区域4 种土地利用类型的土壤为中性和弱碱性土壤 (表1). 其中, 耕地的土壤孔隙度和土壤含水率在4 种土地利用类型中均为最高, 显著高于坑塘水域、仓储用地和居民居住区.

表1 不同土地利用类型的土壤理化性质Tab. 1 Soil environmental factors of different land use types
2.2 测序数据统计与物种鉴定
测序质控结果表明, 4 种土地利用类型23 个样本中共获得100 万条长度为421 ~ 460 bp 的16S rRNA 序列和160 万条长度为201 ~ 360 bp 的ITSrRNA 序列, 用于分析土壤细菌和土壤真菌的多样性格局. 根据97%的序列相似性共鉴定出632 种土壤细菌OTUs 和593 种土壤真菌OTUs. 研究区域中多度最高的土壤细菌OTUs 来自Pseudarthrobacter, 多度最高的土壤真菌OTUs 来自被孢霉属(Mortierella). 4 种土地利用类型中分别有446 ~ 530 种土壤细菌OTUs 和267 ~ 331 种土壤真菌OTUs. 其中, 耕地的细菌OTUs 最多, 真菌OTUs 最少; 居民居住区的真菌OTUs 最多, 细菌OTUs 最少. 土壤细菌和土壤真菌的OTUs 数量在不同土地利用类型间无显著差异(Q> 0.05). Rarefaction 分析结果表明, 各样品稀释曲线的末端均趋于平坦, 说明数据采集充分, 能够准确反映各样品中土壤细菌和土壤真菌的群落组成信息(图1).

图1 稀释曲线图Fig. 1 Rarefaction curve
门水平上, 4 种土地利用类型中土壤细菌和土壤真菌的优势类群相同, 为变形菌门(Proteobacteria,占比39.40% ~ 53.62%)和子囊菌门(Ascomycota, 占比77.61% ~ 95.82%). 但优势类群的相对多度在不同土地利用类型间存在显著差异, 变形菌门的相对多度在耕地中最高且与居民居住区存在差异显著(p= 0.045). 属水平上, 各土地利用类型的优势细菌属为Pseudarthrobacter和Gaiella, 但相对多度在土地利用类型间差异显著.Pseudarthrobacter的相对多度在坑塘水域和居民居住区之间差异显著(p= 0.02). 此外, 仓储用地中Gaiella的相对多度分别与坑塘水域(p= 0.036)和居民居住区(p=0.022)达到显著差异. 而优势土壤真菌属主要为各土地利用类型的特有优势真菌属 (图2).

图2 不同土地利用类型中土壤微生物的群落组成Fig. 2 Community composition of soil microbes across different land use types
2.3 不同土地利用类型中土壤微生物的α 多样性
4 种 土地 利 用类 型中, 土 壤细 菌的Chao1 指数为317 ~ 374, Coverage 指数 为0.937 ~ 0.949,Shannon 指数为4.41 ~ 4.71; 该指数最高的是坑塘水域, 最低的是耕地; 土壤细菌的丰富度、覆盖度和α 多样性在4 种土地利用类型间无显著差异(图3(a)、(c)、(e)). 土壤真菌的Chao1 指数为107 ~157; Coverage 指数在4 种土地利用类型中均达到0.99 以上(0.992 ~ 0.995), Shannon 指数为1.99 ~2.60, 该指数最高的是仓储用地, 最低的是坑塘水域; 土壤真菌的丰富度、覆盖度和α 多样性在4 种土地利用类型间无显著差异(图3(b)、(d)、(f)).

图3 不同土地利用类型中土壤微生物的α 多样性图Fig. 3 α Diversity of soil microbes across different land use types
2.4 不同土地利用类型中土壤微生物的β 多样性
基于Bary-curtis 距离算法的主坐标分析结果显示, 研究区域的土壤细菌和土壤真菌分别聚成了两类(图4). PERMANOVA 结果显示, 土地利用类型与土壤理化指标相比, 对土壤细菌和土壤真菌β 多样性格局的解释度更高(表2). 通过基于相同距离算法的相似性分析(analysis of similarities,ANOSIM), 计算4 种土地利用类型间的微生物群落结构差异. 结果表明, 坑塘水域与耕地、仓储用地、居民居住区这3 种土地利用类型的土壤细菌群落结构差异显著(p< 0.05), 此外, 居民居住区与耕地、仓储用地这两种土地利用类型的土壤细菌群落结构差异显著(p< 0.05) (表3). 土壤真菌的群落结构差异在4 种土地利用类型间表现为坑塘水域与耕地、仓储用地、居民居住区这3 种土地利用类型差异显著(p< 0.05), 仓储用地与居民居住区之间差异显著(p< 0.05) (表3).

图4 不同土地利用类型中土壤微生物的主坐标分析Fig. 4 Principal coordinate analysis of soil microbes across different land use types

表2 置换多因素方差分析结果表Tab. 2 Permutational multivariate analysis of variance

表3 相似性分析结果表Tab. 3 Analysis of similarities (ANOSIM)
多级物种差异判别分析结果显示, 对4 种土地利用类型间差异贡献最大的土壤细菌和土壤真菌为各土地利用类型中的优势土壤细菌和优势土壤真菌, 属水平上, 对耕地中土壤细菌群落结构贡献最大的是地杆菌属(Geobacter,A= 4.60), 对坑塘水域中土壤细菌和土壤真菌群落结构贡献最大的分别是Gaiella(A= 4.37)和腐质霉属(Humicola,A= 4.85), 仓储用地中对土壤细菌和土壤真菌群落结构贡献最大的分别是硫杆菌属(Thiobcaillus,A= 4.38)和柄孢壳属(Zopfiella,A= 5.03), 居民居住区中对土壤细菌和土壤真菌群落结构贡献最大的分别是Dongia(A= 4.32)和头梗霉属(Cephaliophora,A= 4.88).
2.5 土壤理化环境对土壤微生物群落组成和多样性的影响
通过相关性分析明确影响土壤细菌和土壤真菌群落组成的环境因素, 发现土壤细菌和土壤真菌的群落组成差异格局与土壤含水率、土壤孔隙度和土壤酸碱度显著相关(图5). 回归分析的结果发现,土壤酸碱度(细菌R2= 0.000 6,p= 0.92; 真菌R2= 0.066,p= 0.24)、土壤孔隙度(细菌R2= 0.005,p= 0.76; 真菌R2= 0.031,p= 0.42)和土壤含水率(细菌R2= 0.094,p= 0.17; 真菌R2= 0.114,p=0.12)与土壤细菌和土壤真菌α 多样性格局的相关性不显著. 冗余分析和典范对应分析结果显示, 土壤细菌和土壤真菌的β 多样性格局与土壤酸碱度、土壤孔隙度和土壤含水率显著相关 (图6).
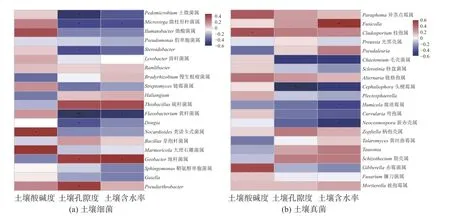
图5 不同土地利用类型中的优势土壤微生物类群与土壤理化指标的相关性分析Fig. 5 Correlation analysis between soil dominant microbes and soil environmental factors across different land types

图6 不同用地类型中土壤微生物β 多样性与土壤理化指标的相关性分析Fig. 6 Correlation analysis between soil microbial β diversity and soil environmental factors across different land types
3 讨 论
研究区域土壤细菌和土壤真菌的群落组成在门水平上表现为: 不同土地利用类型的优势门相同,但相对多度差异显著; 属水平上优势土壤真菌主要是各土地利用类型的特有优势真菌属, 土壤细菌的群落组成格局与门水平一致. 该区域土壤细菌和土壤真菌的β 多样性格局在土地利用类型间呈现显著差异格局. 此外, 土壤细菌和土壤真菌的土地利用类型间差异格局主要由各土地利用类型中的优势土壤细菌和优势土壤真菌贡献. 基于对上述群落组成格局和多样性格局驱动因素的探索, 本研究聚焦于土壤理化指标, 发现该区域土壤细菌和土壤真菌的β 多样性格局和群落组成格局与土壤理化指标(土壤酸碱度、土壤孔隙度和土壤含水率)显著相关.
3.1 土地利用类型对土壤微生物多样性分布格局和群落组成格局的影响
本文聚类所得的土壤细菌OTUs 数目比土壤真菌OTUs 多, 与已发表研究结论相似[37]; 且相对多度最高的土壤细菌和土壤真菌类群与我国北京市的研究工作结论一致, 分别是变形菌门和子囊菌门[38].城乡交错区土壤细菌和土壤真菌的多样性分布格局与已有研究结论一致, 上述两类微生物的α 多样性格局均不受土地利用类型显著影响; 而β 多样性在不同土地利用类型间呈现显著差异格局[27,39]. 造成上述格局的原因可能是各土地利用类型中的土壤基底相同, 故可容纳的微生物物种数相似, 而由于土壤细菌和土壤真菌对干扰的响应较为敏感, 且上海市城乡交错区人类活动强度高[40], 可能会导致不同土地利用类型中微生物的β 多样性差异较大.
土壤基底相同的土地利用类型可容纳相似物种数的微生物, 但各土地利用类型中的微生物结构差异显著, 该差异格局在不同分类学水平上差异表达. 门水平上, 各土地利用类型的优势土壤细菌和优势土壤真菌组成相同, 相对多度呈现的土地利用类型间显著差异格局与已发表研究观点一致, 土地利用类型或土地管理方式的不同会影响土壤微生物的相对多度[41]; 属水平上, 优势土壤真菌为各土地利用类型的特有优势真菌属, 说明该分类学水平上的土壤真菌更敏感, 可能对干扰环境具有更好的指示意义[42].
3.2 土壤理化环境对土壤微生物多样性分布格局和群落组成格局的影响
本文发现土壤细菌和土壤真菌的β 多样性格局与土壤酸碱度、土壤孔隙度和土壤含水率显著相关, 与多项已发表研究结论相似[43-44]. 此外, 有研究表明土壤细菌对土壤酸碱度高度敏感[45-46], 但本文未表现出相似格局. 其原因可能是在研究尺度上土壤基底相同, 土壤酸碱度差异不大(pH 值为7.1 ~8.0). 本文的空间尺度上, 土壤理化环境没有显著影响土壤细菌和土壤真菌的α 多样性格局. 因此, 可能需要更多地考虑空间尺度上的生物因素或时间尺度等[47]. 土壤细菌和土壤真菌的群落物种组成与土壤理化环境显著相关, 可能是因为微生物对土壤理化环境具有较高的敏感度.
3.3 总结与展望
农业活动和城市建设产生的土地利用是改变土壤微生物多样性的主要因素[48-49]. 区域规划和建设活动中, 可以从土地利用类型的角度切入保护城乡交错区土壤微生物多样性, 对被高强度人为干扰的土壤生境采取措施, 尤其是农业用地, 维持土壤健康, 保障粮食安全, 构建可持续发展的土壤生态系统. 对城乡交错区不同土地利用类型土壤微生物多样性的保护工作, 还需要充分考虑土壤酸碱度、土壤孔隙度和土壤含水率的影响, 它们会显著影响土壤微生物的多样性分布格局和群落组成格局. 相对于污染指标, 这类基础指标在城乡建设中往往很容易被忽略, 在污染防控的同时需要关注与土壤环境相关的非污染指标, 保护地下生物多样性, 维持土壤生态系统的功能与服务.
