全基因组DNA甲基化和转录组联合分析鉴定杜梨耐盐相关转录因子
2023-04-10李慧张雨峰李晓刚王中华蔺经常有宏
李慧,张雨峰,2,李晓刚,王中华,蔺经,常有宏
全基因组DNA甲基化和转录组联合分析鉴定杜梨耐盐相关转录因子
李慧1,张雨峰1,2,李晓刚1,王中华1,蔺经1,常有宏1
1江苏省农业科学院果树研究所/江苏省高效园艺作物遗传改良重点实验室,南京 210014;2南京林业大学生物与环境学院,南京 210037
【】鉴定不同杜梨株系根中响应盐胁迫信号的相关转录因子,分析盐胁迫下基因序列DNA甲基化变化与基因表达改变之间的关系,探讨参与调控不同杜梨株系耐盐能力的转录因子成员。【】以杜梨耐盐株系和普通株系为试材,在苗期使用200 mmol∙L-1NaCl对90日龄组培生根苗进行水培处理,以Hoagland营养液为对照。利用火焰石墨炉原子吸收光谱仪测定钠离子含量;利用全基因组DNA甲基化和转录组测序技术从表观遗传修饰和转录调控水平对盐胁迫下转录因子进行生物信息学分析;最后用McrBC-PCR和qPCR对差异转录因子进行验证。【】外源NaCl处理24 h后,杜梨植株中钠离子含量显著增加,其中耐盐株系的增加幅度比普通株系小,为普通株系钠含量的73.1%,但根中积累的钠是普通株系含量的1.1倍;杜梨根中检测到69类共2 682个转录因子的表达,盐胁迫后243个转录因子在两个株系中都发生了差异表达,包括AP2/ERF(37个)、bHLH(19个)、bZIP(7个)、HD-Zip(10个)、MYB(30个)、NAC(18个)、WRKY(8个)和ZFP(23个)等家族成员;盐胁迫后,耐盐株系基因组中转录因子甲基化水平下降,而普通株系转录因子甲基化水平上升,其发生DNA差异甲基化区域主要在基因启动子位置,差异甲基化类型主要为mCHH,占mCG、mCHG、mCHH三种类型总和的93%以上。AP2/ERF、bHLH、DREB、GRAS、GT因子、HB Zip、MYB、NAC、Trihelix和Zinc finger ZFP家族的23个转录因子响应盐胁迫表达量上调而甲基化水平降低,可能参与调节钠在根中的吸收和积累。对部分候选基因的表达模式和启动子区域分别进行实时荧光定量(qPCR)和甲基化依赖型限制性内切酶PCR(McrBC-PCR),验证了生物信息学分析结果。【】盐胁迫后在两个杜梨株系根中均差异表达的转录因子数目为243个,其中8个转录因子(、、、、、、、)DNA序列的甲基化改变与基因转录水平变化呈负相关,研究结果为揭示转录因子参与不同杜梨株系耐盐能力调控的分子机制提供了依据。
杜梨;盐胁迫;转录因子;DNA甲基化;转录组
0 引言
【研究意义】由于极端气候变化和不合理的农业活动,全球约7%面积的土地(超过9亿hm2)受到不同程度的盐渍化危害[1]。耐盐新品种的选育是减少土壤盐渍化对农业生产不良影响的有效途径[2],研究不同物种应对盐胁迫的具体机制是促进耐盐作物育种、合理利用盐渍化土地的首要前提。杜梨是我国第三大果树梨的常用抗逆砧木,用其作砧木嫁接,可以提高梨品种的耐盐能力。发掘杜梨耐盐相关基因,研究该物种的耐盐机制对促进梨生产及盐渍土地的开发利用具有重要意义。【前人研究进展】盐胁迫主要产生渗透胁迫、离子胁迫和次生胁迫,植物在基因水平上进行转录调控是应对上述胁迫的关键步骤,转录因子在应激反应基因的表达中起着关键的调控作用[3-4]。利用微阵列技术分析盐胁迫条件下拟南芥转录组的变化,发现有289个转录因子受盐胁迫诱导上调表达,139个转录因子的表达被抑制[5]。陆地棉根中有123个转录因子基因受盐胁迫诱导表达[6],苜蓿中受盐胁迫诱导而上调表达的基因中20%为转录因子[7]。同一物种中不同耐盐能力的品种之间转录因子对盐胁迫的转录响应也存在差异,如盐胁迫下两个甜瓜品种差异表达的转录因子既表现特异性,也存在部分重叠[8]。除了基因转录调控外,植物还改变全基因组DNA甲基化状态以应对盐胁迫[2],植株基因型是影响盐胁迫下基因组DNA甲基化多态性改变的主要因素[9]。例如:水稻盐耐受品种通过去甲基化来响应盐胁迫,其甲基化水平比盐敏感品种更低[10]。而棉花的盐耐受品种比敏感品种的DNA甲基化水平更高[11]。植物在高盐逆境下DNA甲基化状况的自我调节,是应对盐胁迫、调控基因时空表达所必需的表观遗传适应机制。已有研究发现转录因子通过改变自身基因序列的DNA甲基化状况来响应盐胁迫,例如水稻14个锌指蛋白基因的甲基化状态受盐胁迫诱导改变,从而激活或抑制该类基因的转录,参与调控植株应对高盐逆境[12]。水稻基因组中的基因型特异性表观遗传变化可能是通过改变盐胁迫响应基因的表达网络来感知和响应盐胁迫的重要替代调控机制[13]。而某些转录因子的DNA甲基化对基因表达的激活或抑制在大豆盐胁迫耐受过程起关键作用[14]。【本研究切入点】目前杜梨耐盐研究集中于生理评价和基因表达分析[15-18],该物种中不同转录因子对盐胁迫存在表达响应[19-20],但缺乏盐胁迫下所有转录因子表达情况的全面分析,其基因DNA序列甲基化状况对转录水平的影响也不清楚。【拟解决的关键问题】以耐盐能力不同的2个典型杜梨株系为材料,拟从转录组和全基因组甲基化联合分析入手,系统研究杜梨盐胁迫下转录因子表达调控情况,结合DNA甲基化状态变化情况,鉴定杜梨耐盐相关转录因子,为耐盐品种培育和盐碱地高效利用提供技术参考。
1 材料与方法
试验于2019—2022年在江苏省农业科学院果树研究所进行。
1.1 试验材料处理与钠离子测定
以杜梨耐盐株系(收集于连云港花果山,可耐6‰ NaCl)和普通株系(收集于南京紫金山,仅耐3‰ NaCl)组培苗为试材,经生根培养基诱导生根30 d后,转入温室土培(营养土﹕蛭石3﹕1)90 d,选择生长一致的植株,轻轻洗净根部基质,两种株系各60 株分为两组,一组加入额外添加200 mmol∙L-1NaCl的Hoagland营养液进行胁迫处理,一组加入Hoagland营养液作为对照[21]。处理24 h后分别收集植株全株或单独的根系,去离子水充分冲洗后,全株分为地上部和根系两部分烘干,用于钠离子测定;而新鲜根系经液氮急冻后保存于-80℃超低温冰箱待用。
杜梨地上部和根系经80℃烘干6 h,每0.5 g样品加入25 mL醋酸(1 mol·L-1),90℃振荡消化2 h,过滤定容后于火焰石墨炉原子吸收光谱仪(ZEEnit®700P,德国耶拿)测定钠离子含量,全株的钠离子含量由地上部和根系两部分含量相加。钠标准溶液购自美国默克公司。
1.2 转录组测序与数据分析
使用TRIzol 试剂(Invitrogen)分离和纯化杜梨根总RNA。建库RNA满足浓度>100 ng·μL-1、RIN number>7.0、OD260/280>1.8、总含量>20 μg。NEBNext®Ultra™ RNA Library Prep Kit for Illumina®(NEB)生成测序文库及Illumina HiSeqTM2500双端测序均在南京集思慧远生物技术有限公司进行,每个处理的样品均进行3次生物学重复。
使用FASTQC(http://www.bioinformatics.babraham. ac.uk/projects/fastqc/)读取质量控制指标,NGS QC工具包用于去除接头序列和低质量序列[22]。使用HISAT(v2.1.0)[23]将过滤后的干净序列比对至杜梨参考基因组[24],参数设置为默认标准。获得的比对序列组成bam文件计算FPKM(fragments per kilobases per million reads)值来代表基因表达水平。R包DESeq2[25]用于鉴定差异表达基因,标准为|log2Fold change|>1且FDR<0.05。使用iTAK(1.7a)软件从杜梨基因组的所有基因中鉴定转录因子[26]。
1.3 DNA甲基化测序与数据分析
使用 DNeasy Plant Mini Kit(Qiagen)提取杜梨根DNA,MinElute PCR Cleanup(Qiagen)对DNA样品进行纯化并测定浓度。1 μg基因组DNA经超声波处理机械打断、末端修复、3'端加A碱基、连接甲基化接头后,使用EZ DNA Methylation-Gold™试剂盒(Zymo Research)将DNA转化为亚硫酸氢盐,全基因组重亚硫酸盐测序(whole-genome bisulfite sequencing,WGBS)文库的构建以及Illumina HiSeqTM2500双端测序均在南京集思慧远生物技术有限公司进行,每组样品均进行3次生物学重复。
使用Trim fastp(v0.20.0)[27]对原始测序读数进行质量过滤和接头去除。随后使用Bismark(v0.22.3)- bowtie2(2.3.5.1)[28]将高质量修剪后的读段与杜梨参考基因组[24]进行比对。使用Bismark(命令为—no_ overlap--ignore 5--ignore_r2 5)识别比对读数中的甲基化胞嘧啶,5mC检测的筛选条件为:覆盖度≥4X、FDR<0.05。使用MethylKit(v1.12.0)[29]在R包(v3.6.0)中识别差异甲基化区域(differentially methylated region,DMR),参数为span=1 000、FDR<0.05、C>0.2、CG>0.25、CHG>0.20、CHH>0.1。使用Bedtools(v2.21.0)[30]对DMR进行基因特征注释。
1.4 转录组和甲基化联合分析
使用Bedtools(v2.21.0)在R包(v3.6.0)中注释来自WGBS分析的DMR区域[30]。使用Perl编程语言获得目标基因进行分析,它们是在指定位置与差异甲基化区域(DMR)相交的转录组结果的差异甲基化基因(differentially methylated gene,DMG)。最后,结合iTAK(1.7a)软件鉴定出的转录因子ID号信息确定DMG中包含的转录因子成员[26]。
1.5 McrBC-PCR和qPCR
qPCR 基因特异性引物使用Primer Premier 5.0设计,上海生工合成,引物序列见表1。所有引物均用PCR扩增、电泳和溶解曲线进行测试以保证引物特异性。使用LightCycler®480 II (Roche)进行qPCR,反应体系按Genious 2×SYBR Green Fast qPCR Mix (AB clonal)说明书进行。以为内参,用2-ΔΔCT公式计算相对表达量[16,31]。
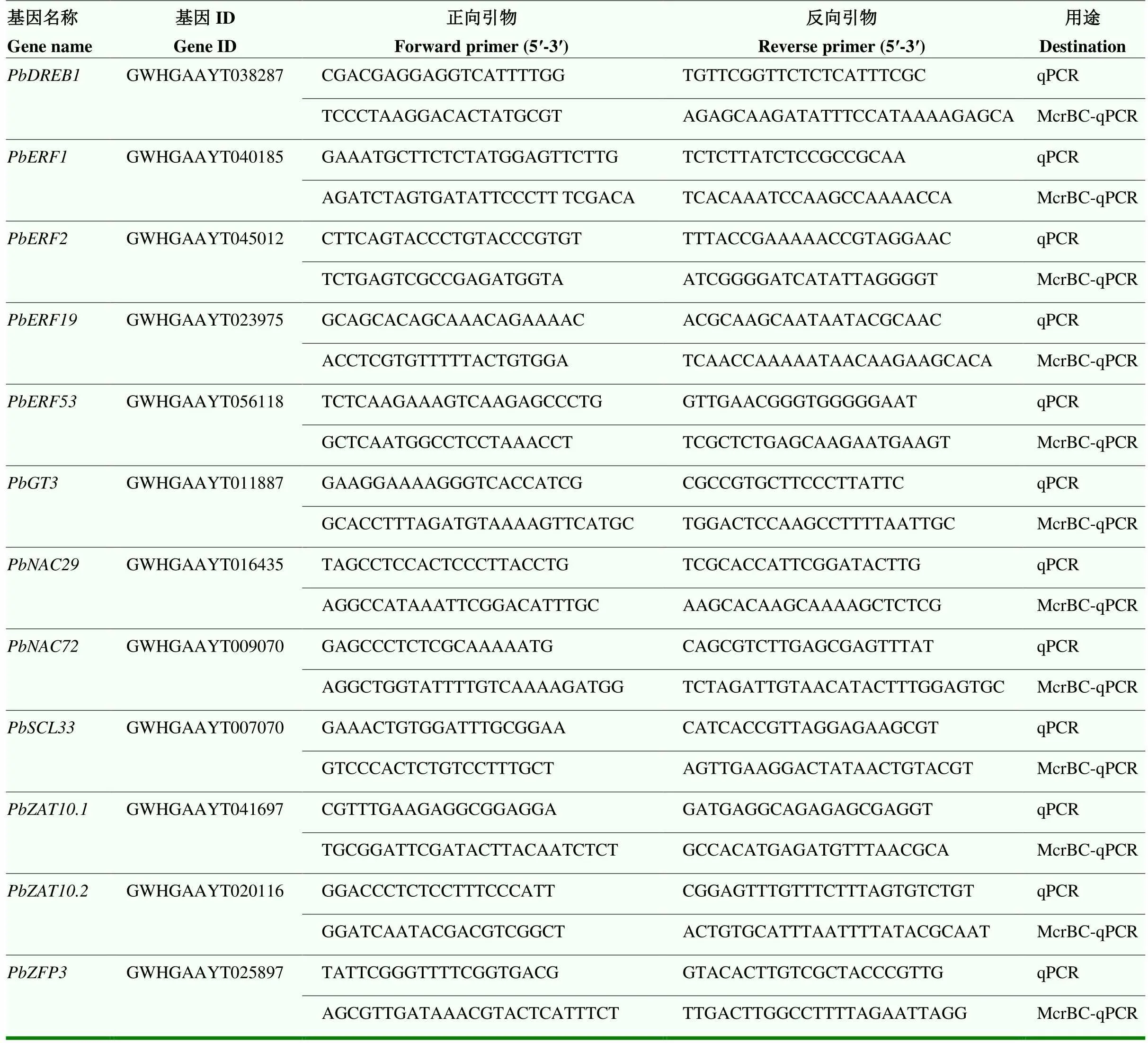
表1 实时荧光定量PCR和McrBC-qPCR引物
用核酸内切酶McrBC(M0272S,New England Biolabs)按照说明书的方法对1 μg的杜梨根基因组DNA进行酶切消化。未加GTP的酶切消化反应作为阴性对照用于标准化分析。甲基化的DNA可以被McrBC消化,PCR结果表现为qPCR信号水平与甲基化水平呈负相关[32]。McrBC qPCR引物设计和合成同qPCR分析,引物序列见表1。
1.6 数据分析
试验数据采用SPSS20进行ANOVA分析,不同处理之间差异采用Duncan检测,数值表示为3个生物学重复的平均值±标准误,不同小写字母表示显著性差异(<0.05)。
2 结果
2.1 不同杜梨株系钠离子含量
在正常生长条件下,耐盐株系和普通株系全株钠离子含量无显著差异,它们根系中钠离子含量占全株的比例相差不大;200 mmol·L-1NaCl处理24 h后,耐盐株系全株钠离子含量是未处理前的3.41倍,根系中钠离子含量占全株的47.09%;普通株系全株钠离子含量是未处理前的4.52倍,根系中钠离子含量仅占全株的35.79%(表2),表明盐胁迫条件下耐盐株系能够将较多的钠离子储存在根中,限制其向上运输,从而达到盐耐受的目的。
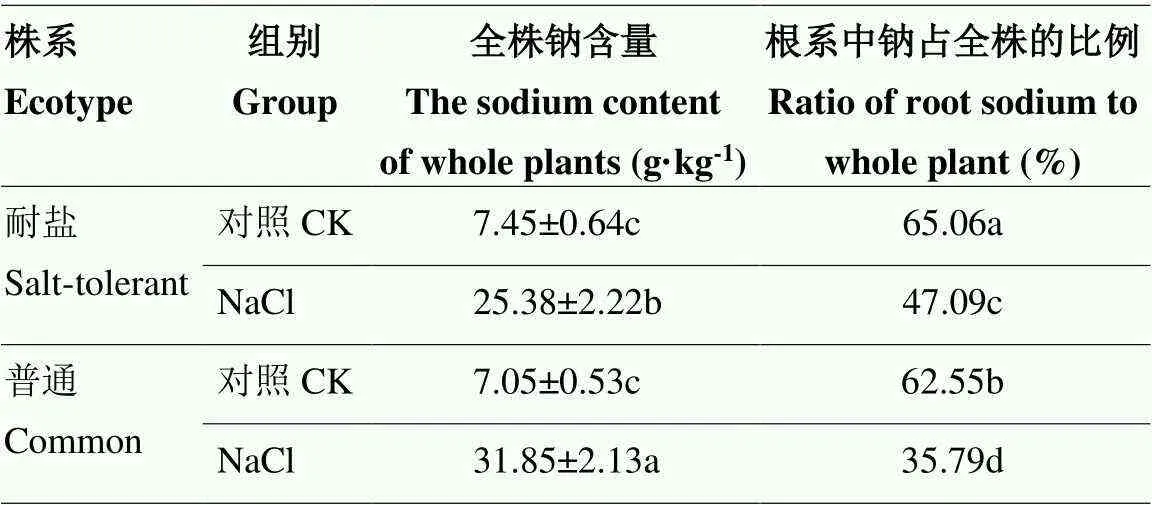
表2 不同杜梨株系盐胁迫后钠离子含量
不同小写字母表示不同处理间差异显著(<0.05)
Different lowercase letters indicate significant difference (<0.05)
2.2 不同杜梨株系表达差异转录因子
从12个杜梨根系样品中分别获得2 069.3—3 025.4万条不同数目的转录组有效序列,Q30≥90.8%,各样品数据量≥6.2 G,共获得96.4 Gb数据供后续分析(NCBI number: PRJNA812627),每个处理的3次生物学重复样品间的一致性为91.1%—98.9%。转录组数据与杜梨参考基因组(http://bigd.big.ac.cn/gwh/ Assembly/505/show)的比对率为84.8%—86.9%。借助iTAK(1.7a)软件从杜梨基因组中鉴定出69类共2 682个转录因子基因(图1)。将200 mmol·L-1NaCl处理24 h后的杜梨耐盐株系或普通株系根转录组数据分别和其对照根转录组数据进行比对分析,分别获得702个和435个差异表达转录因子基因(图2);其中耐盐株系处理24 h后,共计342个转录因子基因表达上调,360个转录因子基因表达下调;普通株系处理24 h后,共计192个转录因子基因表达上调,243个转录因子基因表达下调;耐盐株系和普通株系盐胁迫前后共同的表达差异转录因子基因有243个,包括AP2/ERF(37个)、bHLH(19个)、bZIP(7个)、HD-Zip(10个)、MYB(30个)、NAC(18个)、WRKY(8个)和ZFP(23个)等基因家族成员;其中表达趋势相同的转录因子基因为231个(115个上调,116个下调);表达趋势不同的差异转录因子基因有12个。
2.3 不同杜梨株系转录因子差异甲基化区域
12个杜梨根系样品的WGBS测序原始数据过滤后得到6 411.3—8 305.5万条有效序列,Q30≥91.1%,分别获得19.2—24.9 Gb数据(NCBI number: PRJNA812739),是杜梨参考基因组[24]大小(532.7 Mb,http://bigd.big.ac.cn/gwh/Assembly/505/show)的36.1— 46.8倍,WGBS数据与杜梨参考基因组的比对率为64.4%—72.8%。不同样品之间转录因子相同基因区域的甲基化水平类似,以转录起始位点为界限,基因编码区的甲基化程度最低,基因上游(2 kb)及下游(2 kb)区域甲基化程度较高;盐胁迫后,耐盐株系基因组中转录因子甲基化水平下降,而普通株系转录因子甲基化水平上升;两个株系发生DNA差异甲基化的区域均主要为基因启动子序列(图3)。比较杜梨耐盐株系和普通株系盐胁迫前后WGBS数据,分别获得转录因子基因序列中差异甲基化区域(包括mC、mCG、mCHG、mCHH 4种类型)889和857个,其中mCHH类型的差异甲基化区域占所有差异甲基化区域数目的87.7%和86.0%。耐盐株系盐胁迫处理24 h后,转录因子基因序列中386个差异甲基化区域甲基化水平升高,503个差异甲基化区域的甲基化水平降低;普通株系盐胁迫处理24 h后,转录因子基因序列中569个差异甲基化区域的甲基化水平升高,288个差异甲基化区域的甲基化水平降低(图4、图5)。
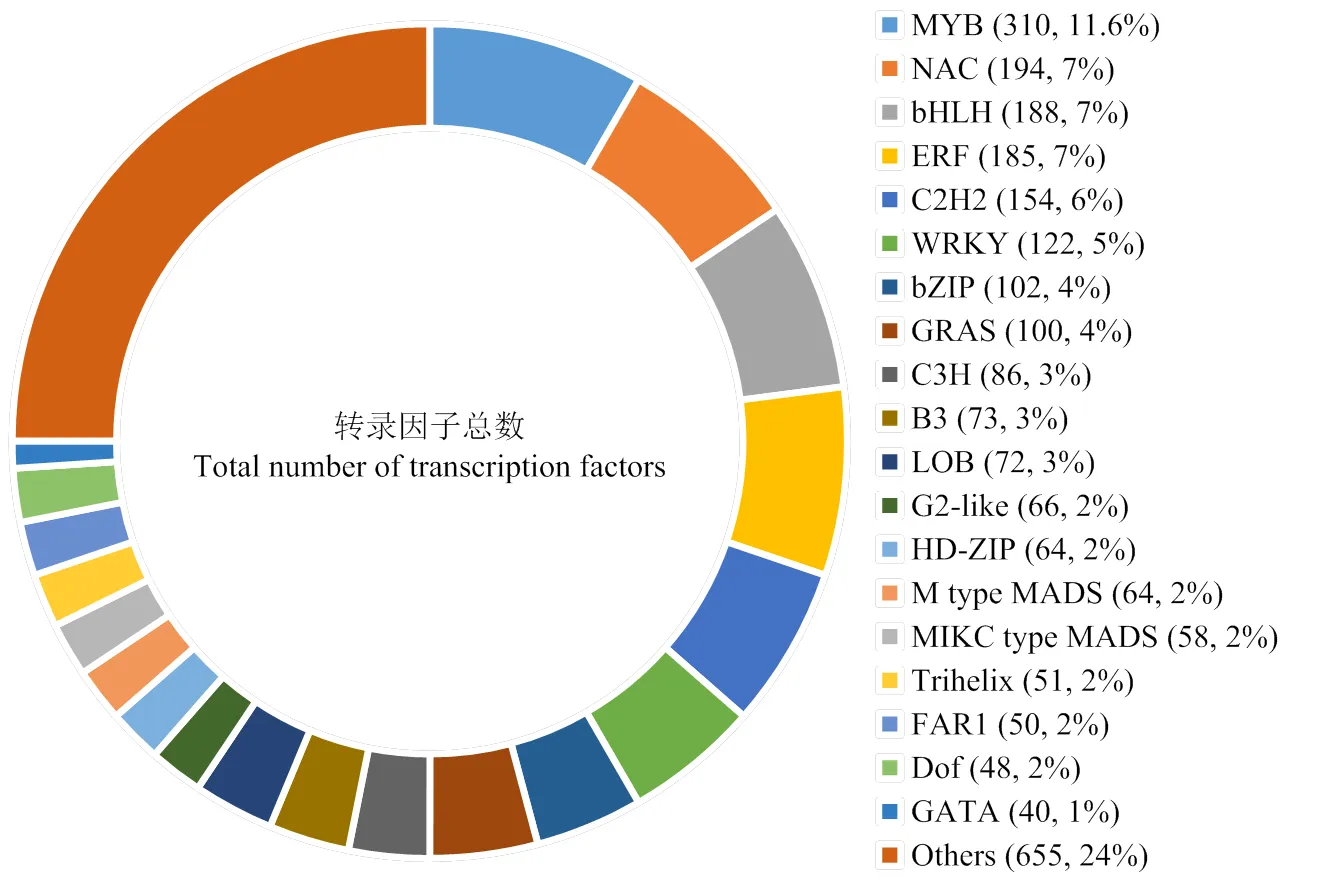
图1 杜梨根中检测到的转录因子种类及比例
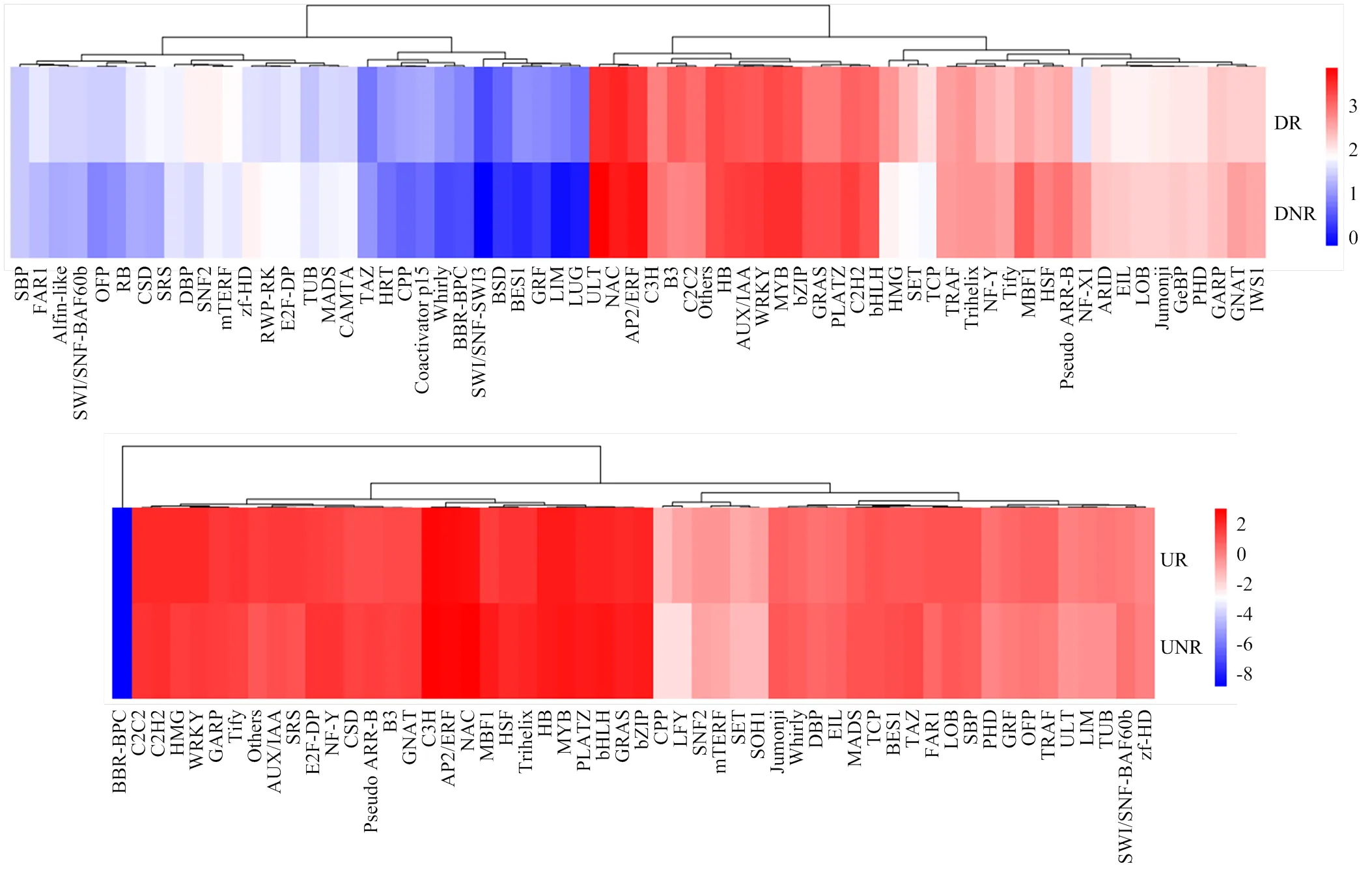
DR:耐盐株系对照根;DNR:耐盐株系200 mmol·L-1 NaCl处理24 h的根;UR:普通株系对照根;UNR:普通株系200 mmol·L-1 NaCl处理24 h的根。下同
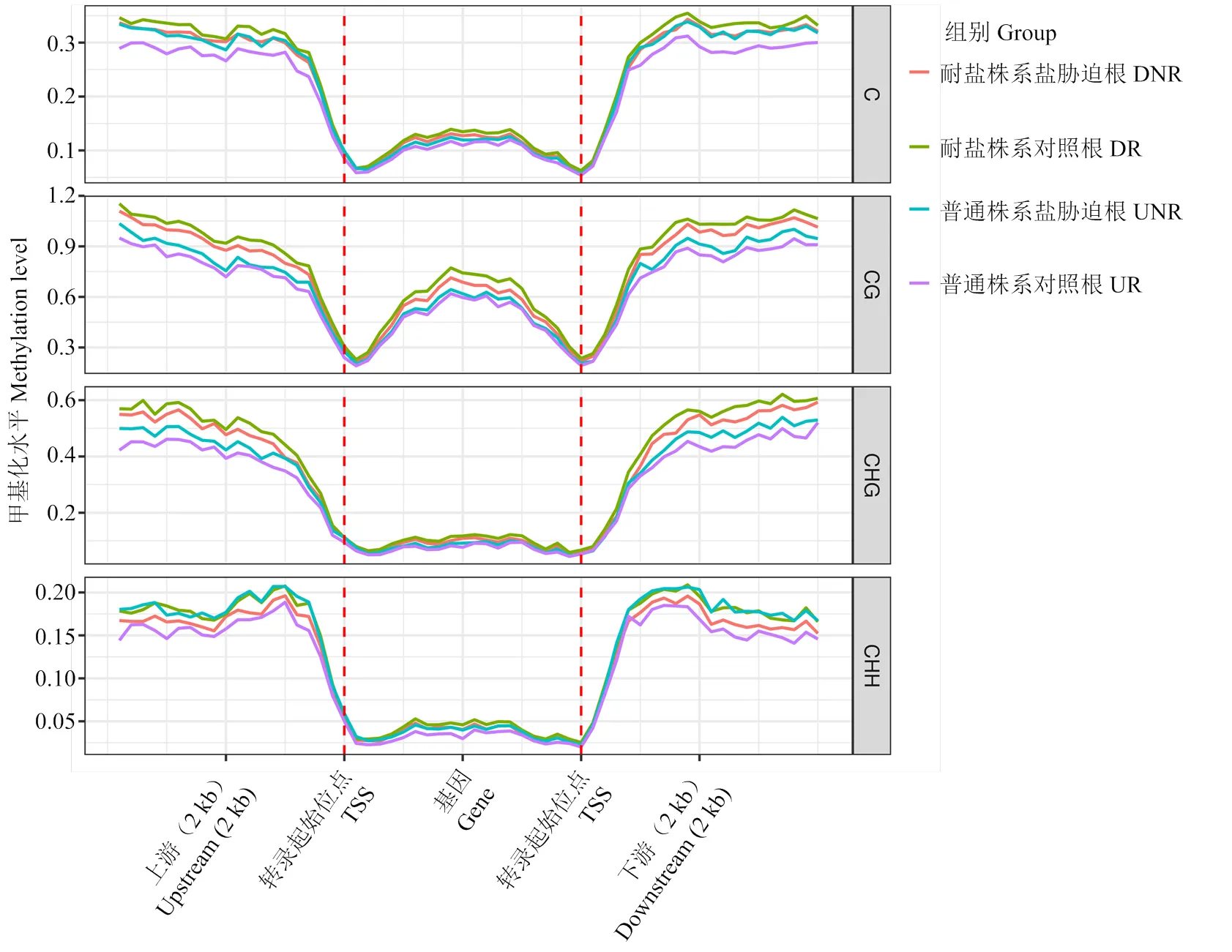
图3 杜梨根中转录因子不同基因区域的甲基化水平
2.4 联合分析与验证
将WGBS和RNA-Seq数据进行联合分析,发现盐胁迫后耐盐株系中同时发生表达差异和甲基化差异的转录因子基因共203个(包括275个差异甲基化区域),其中负相关的转录因子基因为98个,而普通株系中表达差异和甲基化差异相关的转录因
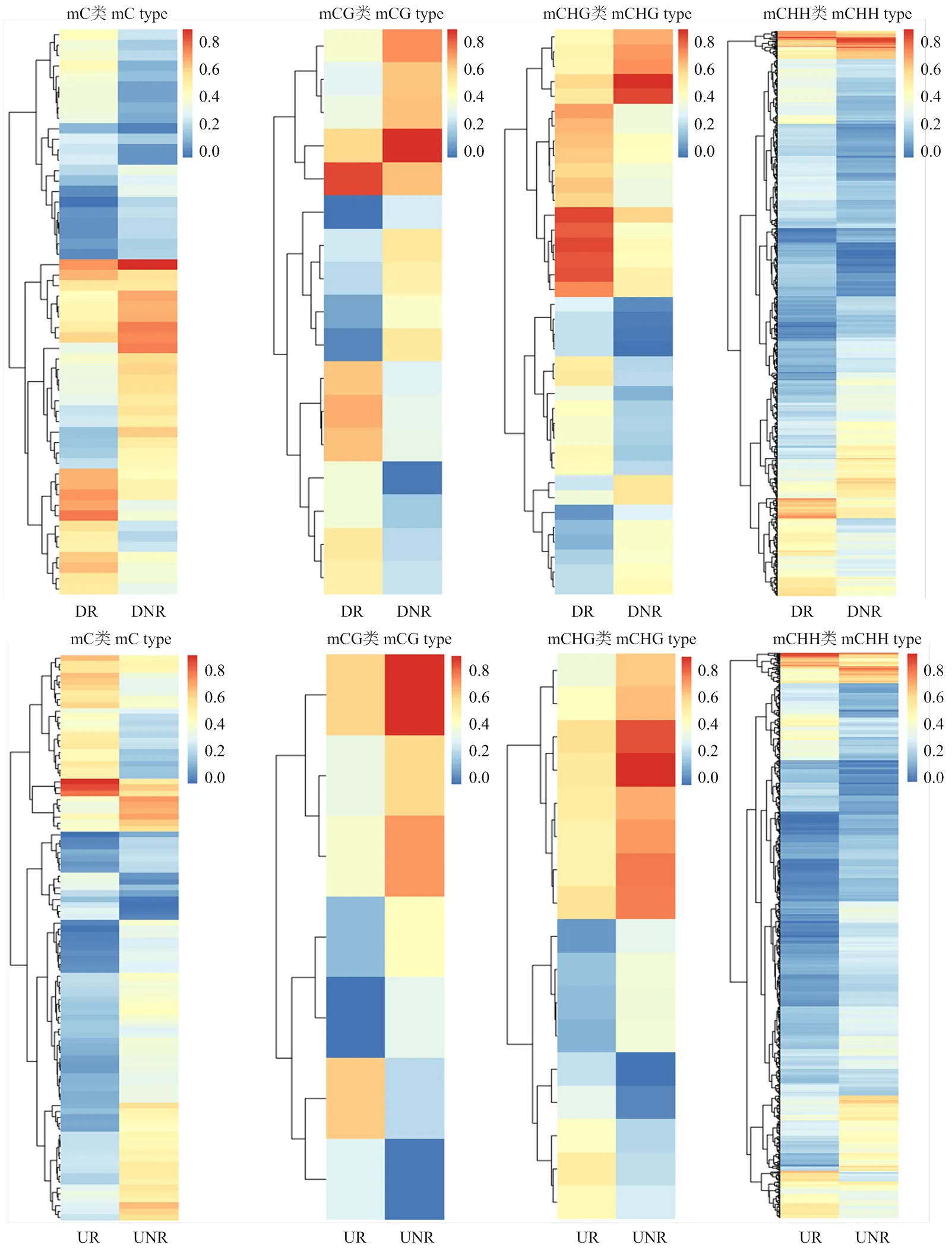
图5 盐胁迫后杜梨不同株系根中在转录因子上的差异甲基化区域热图
子基因共121个(包括178个差异甲基化区域),其中负相关的转录因子基因为60个(表3)。联合分析获得的盐胁迫相关差异转录因子基因中,耐盐株系和普通株系共有的mC类甲基化差异转录因子基因只有1个(GWHGAAYT031490),无相同的mCG类或mCHG类甲基化差异转录因子基因,而它们共有的mCHH类甲基化差异转录因子基因为63个。

表3 联合分析杜梨不同株系盐胁迫后差异转录因子数量
*:基因区域中差异甲基化区域和差异表达基因的交集数量,括号表示基因数量;**:启动子区域中差异甲基化区域和差异表达基因的交集数量,括号表示基因数量
*: The number of intersections between differentially methylated regions and differentially expressed genes in the gene region, the number of genes was showed in the brackets; **: The number of intersections of differentially methylated regions and differentially expressed genes in the promoter region, the number of genes was showed in the brackets
选择盐胁迫后基因转录变化与甲基化状态相关的12个转录因子(IGV软件展示差异甲基化区域,附图1),对它们在两个株系中的表达水平和差异甲基化区域进行qPCR和McrBC-qPCR验证。结果表明,盐胁迫后(GWHGAAYT045012)、(GWHGAAYT011887)、(GWHGAAYT041697)、(GWHGAAYT007070)、(GWHGAAYT038287)在耐盐单株和普通单株中表达水平上升,并且检测区域的甲基化水平下降,其中耐盐单株的变化幅度远大于普通单株;而(GWHGAAYT020116)、(GWHGAAYT056118)、(GWHGAAYT 009070)在耐盐单株和普通单株中表达水平上升,并且检测区域的甲基化水平下降,但是它们在普通单株中表达水平和甲基化水平变化不显著;(GWHGAAYT023975)、(GWHGAAYT016435)在耐盐单株和普通单株中表达水平上升,而检测区域的甲基化水平在耐盐单株中下降、普通单株中上升;(GWHGAAYT040185)、(GWHGAAYT 025897)在耐盐单株和普通单株中表达水平上升,而检测区域的甲基化水平在耐盐单株中无显著变化、在普通单株中下降(图6、图7)。上述结果说明盐胁迫诱导的DNA甲基化变异在不同的耐盐单株中均可调控部分相关转录因子基因(、、、、)的表达变化,而有些响应盐胁迫的差异表达基因(、、)可能并不直接受DNA甲基化所调控,表现为它们的转录水平上升与甲基化变化并不是负相关。在盐胁迫条件下,DNA甲基化如何精确调控特定基因表达,影响杜梨不同株系耐盐能力差异的具体调控机制仍需进一步探究。
3 讨 论
3.1 转录因子参与杜梨耐盐调控过程
前人研究发现,全基因组水平上发生转录改变是植物受到盐胁迫后的普遍反应,而转录因子作为RNA转录网络调控的枢纽,它们在高盐逆境条件下表达水平的变化尤为常见[3-4]。例如拟南芥、陆地棉和苜蓿在盐胁迫条件下,都检测到大量转录因子的表达量提高或降低[5-7]。本研究结果表明,杜梨根系在盐胁迫后16.2%—26.2%的转录因子基因受诱导上调或下调表达。除了基因转录调控之外,全基因组DNA甲基化状态改变也是应对盐胁迫的重要手段,植物可通过DNA低甲基化或高甲基化来调节基因表达以适应高盐逆境[2,33-35],NaCl处理后,杜梨两个株系根的全基因组DNA甲基化水平均发生了变化[21],表明该物种也存在应对盐胁迫逆境的表观遗传调控机制。已有研究发现转录因子通过改变自身基因序列的DNA甲基化状况来响应盐胁迫,比如水稻、大豆和苜蓿等植物中AP2/ERF、NAC、ZFP转录因子家族成员可改变自身序列的DNA甲基化水平来激活或抑制基因表达,从而参与盐胁迫应答调控,在盐耐受过程起关键作用[12-14,36]。本研究发现,杜梨根系中转录因子基因DNA序列中差异甲基化区域(包括mC、mCG、mCHG、mCHH四种类型)为857—889个,同时发生表达差异和甲基化差异的转录因子基因为121—203个,包括AP2/ERF、bHLH、NAC、MYB、WRKY等家族成员,并且它们的转录水平受盐胁迫影响,其中23个转录因子响应盐胁迫表达量上调而甲基化水平降低,可能参与调节钠在根中的吸收和积累,表明杜梨与其他植物类似,其根系中的转录因子也可通过改变自身基因序列DNA甲基化水平来调节转录情况,从而参与植株的耐盐调控过程。
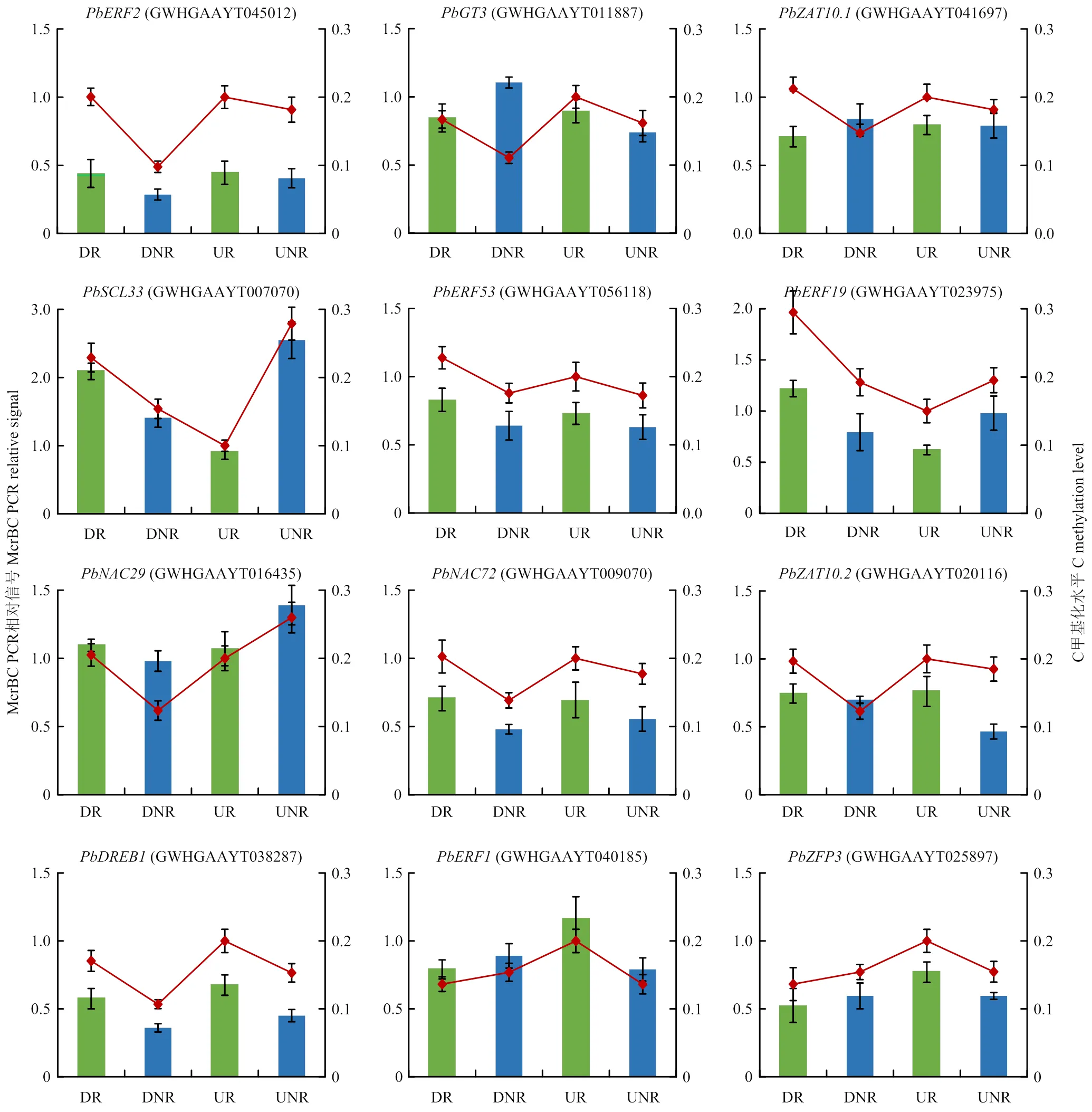
图7 盐胁迫下杜梨不同株系根中转录因子DNA甲基化水平和差异甲基化区域验证
3.2 杜梨响应盐胁迫存在基因型特异性
植物应对高盐胁迫的复杂过程涉及胁迫相关基因的高效有序表达,离不开体内各类转录因子的有效调控[5-8],同一物种中不同耐盐能力的品种之间对盐害的耐受能力主要与基因对胁迫的响应程度相关[37],例如盐胁迫下两个甜瓜品种差异表达的转录因子既表现特异性,也存在部分重叠[8]。杜梨耐盐株系和普通株系盐胁迫前后差异表达的转录因子也存在相同特征,各有特异表达的成员,也有部分重叠的成员,某一具体转录因子基因在杜梨盐胁迫过程所起的作用需要进一步试验。此外,转录因子在高盐逆境条件下表达水平的变化往往伴随着基因DNA序列的甲基化模式改变[5-8],水稻、大豆和苜蓿等植物中AP2/ERF、NAC、ZFP转录因子家族成员在盐胁迫下DNA甲基化改变和转录水平变化与品种特性密切相关,同一物种不同基因型之间存在特异性的表观遗传变化,从而在不同程度上改变盐胁迫响应基因的表达网络来感知和响应盐胁迫,最后表现为植株对盐胁迫的不同耐受能力[12-14,36]。NaCl处理后,杜梨两个株系根的全基因组DNA甲基化水平均发生了变化,普通单株整体甲基化水平上升,耐盐株系通过去甲基化来响应盐胁迫,表现为全基因组甲基化水平下降[21]。本研究发现杜梨耐盐单株根系比普通单株根系DNA甲基化程度低,可能与其能在根系中积累更多的钠离子,从而更好地适应高盐逆境相关。此外,DNA甲基化对基因表达的影响因组织类型、甲基化序列背景、基因间区和基因体内甲基化区域而异,启动子区域的DNA甲基化通常会导致基因表达量下降[38]。本研究中杜梨耐盐株系基因组中转录因子DNA甲基化水平下降,而普通株系转录因子DNA甲基化水平上升;两个株系发生DNA差异甲基化的区域主要为转录因子基因的启动子序列,而前者的转录因子表达水平普遍高于后者,与上述结论相符。杜梨耐盐单株和普通单株中筛选获得共有的盐胁迫相关差异转录因子基因64个,包括AP2/ERF、NAC、ZFP等家族成员,这些基因的表达水平和启动子序列DNA甲基化水平密切相关,但是它们在两个株系中的表现有所差异。表明杜梨在应对盐胁迫时不同株系的根系中转录因子基因的表达情况和DNA甲基化模式不尽相同,该物种的逆境表观遗传调控机制存在基因型特征,而某一具体的转录因子成员如何通过DNA甲基化来调控基因转录的详细过程,进而参与植株适应高盐逆境的表观遗传机制仍需进一步验证。
4 结论
盐胁迫后,杜梨不同耐盐株系基因组中转录因子总体DNA甲基化变化具有基因型特异性,但是两个株系DNA差异甲基化区域均主要在基因启动子位置,差异甲基化类型主要为mCHH。盐胁迫下杜梨根中差异表达基因涉及243个转录因子,其中8个转录因子DNA序列的甲基化改变与转录变化呈负相关,可能参与调控杜梨不同株系耐盐能力差异的分子机制。
[1] LI J G, PU L J, HAN M F, ZHU M, ZHANG R S, XIANG Y Z. Soil salinization research in China: Advances and prospects. Journal of Geographical Sciences, 2014, 24(5): 943-960.
[2] FANG S M, HOU X, LIANG X L. Response mechanisms of plants under saline-alkali stress. Frontiers in Plant Science, 2021, 12: 667458.
[3] YANG Y Q, GUO Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. The New Phytologist, 2018, 217(2): 523-539.
[4] GOLLDACK D, LÜKING I, YANG O. Plant tolerance to drought and salinity: Stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Reports, 2011, 30(8): 1383-1391.
[5] JIANG Y Q, DEYHOLOS M K. Comprehensive transcriptional profiling of NaCl-stressedroots reveals novel classes of responsive genes. BMC Plant Biology, 2006, 6: 25.
[6] GUO J Y, SHI G Y, GUO X Y, ZHANG L W, XU W Y, WANG Y M, SU Z, HUA J P. Transcriptome analysis reveals that distinct metabolic pathways operate in salt-tolerant and salt-sensitive upland cotton varieties subjected to salinity stress. Plant Science, 2015, 238: 33-45.
[7] GRUBER V, BLANCHET S, DIET A, ZAHAF O, BOUALEM A, KAKAR K, ALUNNI B, UDVARDI M, FRUGIER F, CRESPI M. Identification of transcription factors involved in root apex responses to salt stress in. Molecular Genetics and Genomics, 2009, 281(1): 55-66.
[8] 陈嘉贝, 张芙蓉, 黄丹枫, 张利达, 张屹东. 盐胁迫下两个甜瓜品种转录因子的转录组分析. 植物生理学报, 2014, 50(2): 150-158.
CHEN J B, ZHANG F R, HUANG D F, ZHANG L D, ZHANG Y D. Transcriptome analysis of transcription factors in two melon (L.) cultivars under salt stress. Plant Physiology Journal, 2014, 50(2): 150-158. (in Chinese)
[9] ZHANG H M, LANG Z B, ZHU J K. Dynamics and function of DNA methylation in plants. Nature Reviews Molecular Cell Biology, 2018, 19(8): 489-506.
[10] FERREIRA L J, AZEVEDO V, MAROCO J, OLIVEIRA M M, SANTOS A P. Salt tolerant and sensitive rice varieties display differential methylome flexibility under salt stress. PLoS ONE, 2015, 10(5): e0124060.
[11] WANG B H, FU R, ZHANG M, DING Z Q, CHANG L, ZHU X Y, WANG Y F, FAN B X, YE W W, YUAN Y L. Analysis of methylation-sensitive amplified polymorphism in different cotton accessions under salt stress based on capillary electrophoresis. Genes & Genomics, 2015, 37(8): 713-724.
[12] AHMAD F, FARMAN K, WASEEM M, RANA R M, NAWAZ M A, REHMAN H M, ABBAS T, BALOCH F S, AKREM A, HUANG J, ZHANG H S. Genome-wide identification, classification, expression profiling and DNA methylation (5mC) analysis of stress-responsive ZFP transcription factors in rice (L.). Gene, 2019, 718: 144018.
[13] KARAN R, DELEON T, BIRADAR H, SUBUDHI P K. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE, 2012, 7(6): e40203.
[14] SONG Y G, JI D D, LI S, WANG P, LI Q, XIANG F N. The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS ONE, 2012, 7(7): e41274.
[15] LI H, LIN J, YANG Q S, LI X G, CHANG Y H. Comprehensive analysis of differentially expressed genes under salt stress in pear () using RNA-Seq. Plant Growth Regulation, 2017, 82(3): 409-420.
[16] LI H, LIU W, YANG Q S, LIN J, CHANG Y H. Isolation and comparative analysis of two Na+/H+antiportergenes from. Plant Molecular Biology Reporter, 2018, 36(3): 439-450.
[17] WU Q S, ZOU Y N. Adaptive responses of birch-leaved pear () seedlings to salinity stress. Notulae Botanicae Horti Agrobotanici Cluj-Napoca, 2009, 37(1): 133-138.
[18] YU X J, SHI P J, HUI C, MIAO L F, LIU C L, ZHANG Q Y, FENG C N. Effects of salt stress on the leaf shape and scaling ofbunge. Symmetry, 2019, 11(8): 991.
[19] LIN L K, YUAN K L, HUANG Y D, DONG H Z, QIAO Q H, XING C H, HUANG X S, ZHANG S L. A WRKY transcription factorfromfunctions positively in salt tolerance and modulating organic acid accumulation by regulatingexpression. Environmental and Experimental Botany, 2022, 196: 104782.
[20] LIU C X, XU X Y, KAN J L, CHENG Z M, CHANG Y H, LIN J, LI H. Genome-wide analysis of the C3H zinc finger family reveals its functions in salt stress responses of. PeerJ, 2020, 8: e9328.
[21] LI H, ZHANG Y F, ZHOU X Y, LIN J, LIU C X, LI X G, CHANG Y H. Single-base resolution methylome of different ecotype fromreveals epigenomic changes in response to salt stress. Scientia Horticulturae, 2022, 306: 111437.
[22] PATEL R K, JAIN M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE, 2012, 7(2): e30619.
[23] KIM D, LANGMEAD B, SALZBERG S L. HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 2015, 12(4): 357-360.
[24] DONG X G, WANG Z, TIAN L M, ZHANG Y, QI D, HUO H L, XU J Y, LI Z, LIAO R, SHI M, ALI WAHOCHO S, LIU C, ZHANG S M, TIAN Z X, CAO Y F.assembly of a wild pear() genome. Plant Biotechnology Journal, 2020, 18(2): 581-595.
[25] LOVE M I, HUBER W, ANDERS S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 2014, 15(12): 550.
[26] ZHENG Y, JIAO C, SUN H H, ROSLI H G, POMBO M A, ZHANG P F, BANF M, DAI X B, MARTIN G B, GIOVANNONI J J, ZHAO P X, RHEE S Y, FEI Z J. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Molecular Plant, 2016, 9(12): 1667-1670.
[27] CHEN S F, ZHOU Y Q, CHEN Y R, GU J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 2018, 34(17): i884-i890.
[28] KRUEGER F, ANDREWS S R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 2011, 27(11): 1571-1572.
[29] AKALIN A, KORMAKSSON M, LI S, GARRETT-BAKELMAN F E, FIGUEROA M E, MELNICK A, MASON C E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology, 2012, 13(10): R87.
[30] QUINLAN A R, HALL I M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 2010, 26(6): 841-842.
[31] LIVAK K J, SCHMITTGEN T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCTmethod. Methods, 2001, 25(4): 402-408.
[32] MAO H D, WANG H W, LIU S X, LI Z G, YANG X H, YAN J B, LI J S, TRAN L S P, QIN F. A transposable element in agene is associated with drought tolerance in maize seedlings. Nature Communications, 2015, 6(1): 8326.
[33] VIGGIANO L, DE PINTO M C. Dynamic DNA methylation patterns in stress response//RAJEWSKY N, JURGA S, BARCISZEWSKI J. Plant Epigenetics. Cham, Switzerland: Springer, 2017: 281-302.
[34] FINNEGAN E J, PEACOCK W J, DENNIS E S. Reduced DNA methylation inresults in abnormal plant development. Proceedings of the National Academy of Sciences of the United States of America, 1996, 93(16): 8449-8454.
[35] AL-HARRASI I, AL-YAHYAI R, YAISH M W. Differential DNA methylation and transcription profiles in date palm roots exposed to salinity. PLoS ONE, 2018, 13(1): e0191492.
[36] YAISH M W, AL-LAWATI A, AL-HARRASI I, PATANKAR H V. Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (). BMC Genomics, 2018, 19(1): 78.
[37] 潘教文, 李臻, 王庆国, 管延安, 李小波, 戴绍军, 丁国华, 刘炜. NaCl处理谷子萌发期种子的转录组学分析. 中国农业科学, 2019, 52(22): 3964-3976.doi: 10.3864/j.issn.0578-1752.2019.22. 003.
PAN J W, LI Z, WANG Q G, GUAN Y A, LI X B, DAI S J, DING G H, LIU W. Transcriptomics analysis of Na Cl response in foxtail millet (L.) seeds at germination stage. Scientia Agricultura Sinica, 2019, 52(22): 3964-3976. doi: 10.3864/j.issn.0578-1752.2019. 22.003. (in Chinese)
[38] WANG J, MAROWSKY N C, FAN C Z. Divergence of gene body DNA methylation and evolution of plant duplicate genes. PLoS ONE, 2014, 9(10): e110357.
Identification of Salt-Tolerant Transcription Factors in the Roots ofby the Association Analysis of Genome-Wide DNA Methylation and Transcriptome

1Institute of Pomology, Jiangsu Academy of Agricultural Sciences/Jiangsu Key Laboratory for Horticultural Crop Genetic Improvement, Nanjing 210014;2College of Biology and the Environment, Nanjing Forestry University, Nanjing 210037
【】Here, two ecotypes offrom Huaguo Mountain, Lianyungang (the salt-tolerant ecotype, D) and Purple Mountain, Nanjing (the common ecotype, U) were collected for this research. The purpose of this study was to analyze the role of transcription factor genes in the roots of two ecotypes ofdiffering in terms of salt stress. Transcription factors involving in the regulation of the salt tolerance of differentecotypes were identified on the grounds of differential expression under salt stress and the relationship between the methylation status and the relative expression level of relevant tolerance genes after exposure to salt stress was investigated. 【】The 90-day-oldseedlings were grown hydroponically in Hoagland’s nutrient solution supplemented with 200 mmol∙L-1NaCl, with seedlings grown in Hoagland’s nutrient solution as the control. The sodium ion content in the tissues was determined by flame graphite furnace atomic absorption spectrometry. Whole-genome DNA methylation analysis and transcriptome sequencing were performed on three replicates for the following four root samples: ecotype D and ecotype U, each grown in the presence or absence of salt stress. Bioinformatics analysis of transcription factor gene expression under salt stress at the levels of transcriptional regulation and epigenetic methylation were carried out using transcriptome sequencing data and whole-genome DNA methylation results, respectively. Then, McrBC-PCR and real-time fluorescence quantitative PCR (qPCR)were used to confirm the levels of methylation and transcription of differential transcription factor genes. 【】After exogenous NaCl treatment for 24 h, the concentration of sodium ions inroots increased significantly, with the increase in sodium ion concentration in the salt-tolerant ecotype being significantly less than that in the common ecotype. In the whole seedling, the final salt concentration of tolerant ecotype was only 73.1% of that of the common ecotype. Whereas, in the roots, the sodium content of the salt-tolerant ecotype was 1.1 times of that in the common ecotype. These results indicated that the salt-tolerant ecotype could store more sodium ions in roots and limit their upward transport after salt stress. A total of 2 682 transcription factor (TF) genes from 69 gene families were detected in roots. Among them, 243 TF genes displayed differential expression in response to salt stress, including 37 AP2/ERF, 19 bHLH, 7 bZIP, 10 HD-Zip, 30 MYB, 18 NAC, 8 WRKY, and 23 ZFP family genes. The global methylation level of transcription factor genes in the genome of the salt-tolerant rootstock ecotype decreased, whereas the overall methylation level of these genes in the common ecotype increased after exposure to 200 mmol∙L-1NaCl. The differentially methylated regions in both ecotypes were mainly in the position of gene promoters, with the type of differentially methylated sequences being mostly mCHH, constituting more than 93% of the sum of all three types of methylated sequences. The expression levels of twenty-three transcription factor genes, which belonged to the AP2/ERF, bHLH, DREB, GRAS, GT factor, HB Zip, MYB, NAC, Trihelix, and zinc-finger ZFP gene families, were upregulated, and their methylation levels were downregulated in both two ecotypes in response to salt stress. These genes may be involved in the regulation of sodium uptake and accumulation in roots under salt stress. The expression patterns and promoter methylation of representative candidate genes identified by bioinformatics analysis were confirmed by qPCR and McrBC-qPCR.【】The differentially expressed genes in roots ofunder salt stress included 243 transcription factor genes in both ecotypes. The methylation changes in DNA sequences in eight transcription factor genes (,,,,,,, and) were correlated with their transcriptional activity. Our results provided preliminary experimental evidence for supporting a relationship between promoter DNA methylation and expression of TF genes inin response to salt stress as part of the molecular role of TFs involved in the regulation of salt tolerance among differentecotypes, which would increase our understanding of the role of epigenetics in the response of woody trees to abiotic stress.
;salt stress; transcription factors; DNA methylation; transcriptome

2022-05-05;
2023-02-01
江苏省自然科学基金(BK20191238)、江苏现代农业(梨)产业技术体系项目[ATS(2021)436]、国家自然科学基金面上项目(31772287)
通信作者李慧,E-mail:lihui7904@163.com
(责任编辑 赵伶俐)
