抗体介导偶联药物开发中的几个关键点
2023-02-28辛英豪
辛英豪
荣昌生物制药(烟台)股份有限公司
王丽丽
荣昌生物制药(烟台)股份有限公司
李明洋
荣昌生物制药(烟台)股份有限公司
徐霖
荣昌生物制药(烟台)股份有限公司
于占娇
荣昌生物制药(烟台)股份有限公司
李元浩*
荣昌生物制药(烟台)股份有限公司
抗体偶联药物(antibody drug conjugate,ADC)及 核素偶联药物(radionuclide drug conjugate,RDC)是近年来获得广泛关注的靶向生物药,在抗肿瘤治疗中展现了较高的应用价值。此类药物将细胞表达的生物大分子与小分子毒素或者放射性核素通过连接子偶联成为新药。ADC 及RDC 的结构相对复杂,其体内安全性及有效性的影响因素也不同于一般抗体或化药分子。本文讨论了ADC 的开发风险、临床毒性、内吞和药效等,并分析了RDC 的开发进展和独特优势。
1 靶向药物的起源及靶向治疗
自DNA 双螺旋结构破解后,近代生物学的发展在几十年内突飞猛进,跨入了分子时代。随着对疾病本质的深入认识,在疾病干预及药物研发上也不断提高,于20 世纪末开启了针对疾病分子机制中相关靶点的靶向药物研发。
传统药物开发的典型模式可以用针对传染病的抗生素类药物及针对癌症的细胞毒类药物为示范,都是利用体外细胞模型及体内动物疾病模型开展自然界及人工合成物质的有效性及安全性筛选,有成药潜力的物质经进一步优化并完成生产及制剂的工艺开发和质量保障等工作,经药品监管部门的审评审批,最终获批上市用于疾病治疗。在这些传统药物的开发过程中,对药物的作用靶点与机制知之甚少,临床应用中常出现攻击疾病细胞之外的部分正常细胞,使得大部分药物存在一定不良反应。
分子靶向药物是针对特定疾病中与疾病关系密切的靶点开发的药物,与靶点有特异性作用关系,对靶点阳性细胞产生作用,而对靶点阴性细胞不产生作用,从而减少对正常组织和细胞的伤害。靶向药物开发首先在肿瘤药物中获得突破,1998年,美国食品药品监督管理局(FDA)批准了抗人表皮生长因子受体-2(HER2)的人源化抗体trastuzumab(曲妥珠单抗,商品名Herceptin/赫赛汀),成为第一个获准上市的靶向大分子生物药。2001年,另一个靶向药物imatinib(伊马替尼,商品名Gleevec/格列卫)获批上市,成为首个获批上市的靶向小分子化药。伊马替尼是一种酪氨酸激酶抑制剂,靶向因染色体易位而形成的BCR/ABL融合基因,可阻断相关蛋白激酶的作用而抑制肿瘤细胞中的信号传导,临床用于治疗慢性髓性白血病(chronic myelogenous leukemia,CML)等癌症。在伊马替尼早期研发中,结合对靶点激酶的特异性作用,很快锁定其分子结构骨架并进行构效优化,在CML患者来源的细胞及肿瘤模型中获得明确的与靶点相关的体外和体内疗效,于1998年进入临床开发,1999年获得快速通道(fasttrack)资质,2000年进入关键临床阶段,2001年成功获批上市用于治疗CML。
曲妥珠单抗和伊马替尼的相继成功标志着靶向药物时代的开启,引领药物研发进入深层次解读疾病生物学本质、开拓及验证相关靶点、开展靶点聚焦的药物研发及成药性优化等领域,同时推进后续的工艺、质检、临床前和临床开发等。到2017年,已有667 个人类来源的独特蛋白及189 个致病微生物来源的病原蛋白成为药物开发的靶点,其中部分靶点蛋白为G 蛋白偶联受体类(12%)、离子通道类(19%)、蛋白激酶类(10%)等[1]。这些靶点分布于不同人类疾病,均已开展了小分子化药及大分子生物药的研发工作。在Santos 的分析中,人类来源的667 个靶点有540 个开展了小分子药物研发,146 个开展了大分子药物研发,其中部分靶点同时进行着小分子及大分子的药物研发。针对人类靶点开发成功的1104 种药物,有999 种小分子化药以及105 种大分子生物药。针对189 个致病微生物来源的病原靶点,大部分致力于小分子化药的研发,只有7个靶点开展了大分子药物的研发。短短20年,靶向药物的研发取得了亮眼的成绩。
靶向药物的开发针对药物对正常细胞的安全风险作出革命性的改变,加强了药物对靶向细胞的选择性,提高了药物的治疗窗(therapeutic window)。靶向治疗策略在多种治疗方法中的应用及发展,使医疗实现跨越式的进步。表1 总结了部分肿瘤治疗方法的作用机制及对周围细胞的安全性,分析了药物对正常组织的损伤。

表1 部分肿瘤治疗方法的作用机制及对周围细胞的安全性
2 ADC 概况
靶向治疗药物开发中非常值得关注的是结合肿瘤靶点抗原的特异性抗体和小分子细胞毒性药物共同组成的ADC。ADC 是一种靶向治疗癌症的药物,它将特异性结合肿瘤抗原的抗体作为制导成分,以杀死肿瘤细胞的强效毒素作为效应弹头,组成高效靶向生物导弹。抗体将弹头直接引导到肿瘤细胞,避免进入非靶细胞以减少不良反应。至2023年,已有超过15 种ADC 被批准用于治疗癌症,还有多种ADC 正在进行临床试验。表2 是目前上市的ADC 简介。
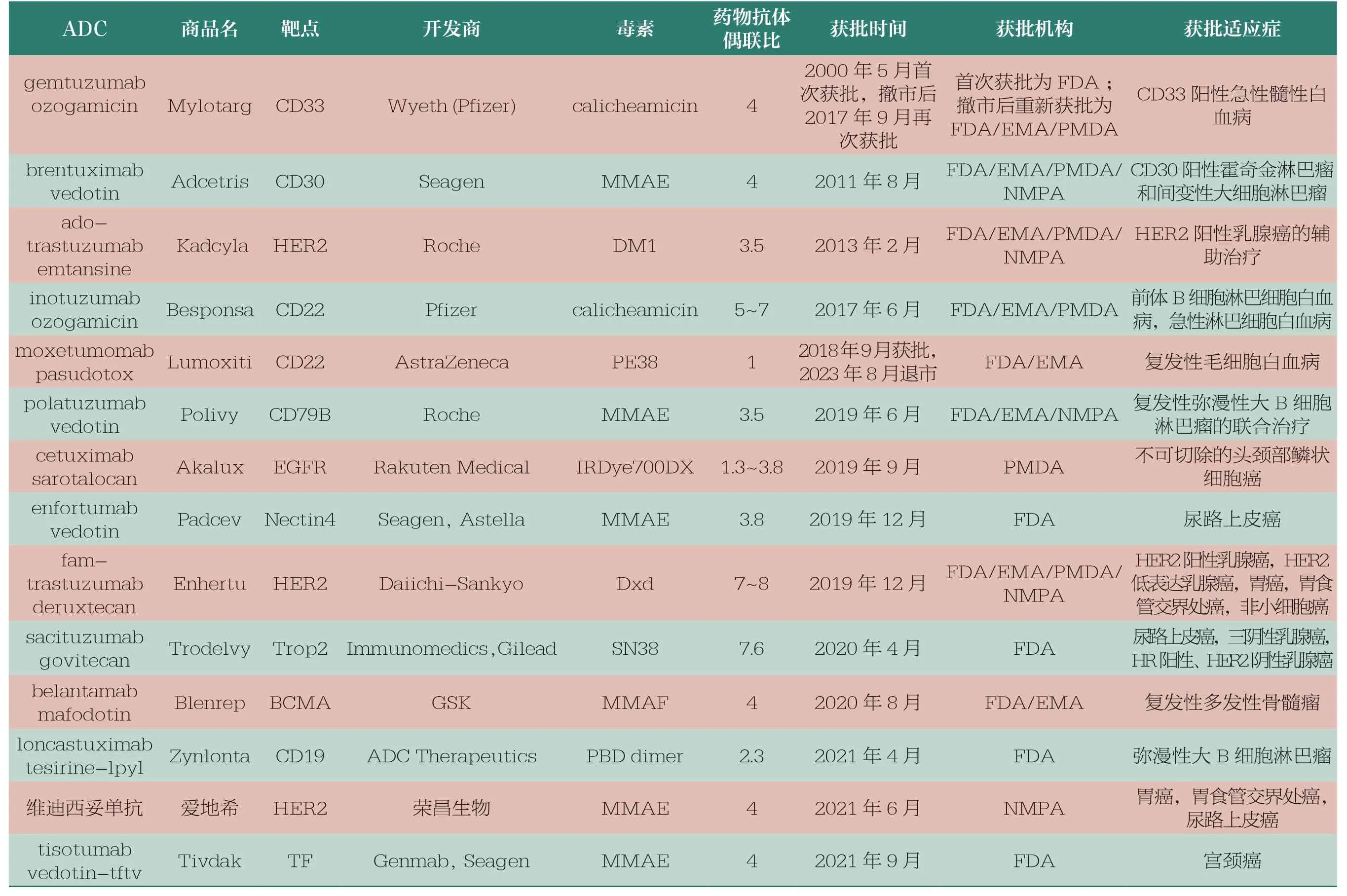
表2 截至2023年初国内外已批准上市的ADC
ADC 近年来在我国的开发异常火热。表3 列出了部分有国内外转让权益的ADC 研发项目,可以客观地反映国内药企在这一研发领域的生命力,也体现了我国生物医药的创新活力和对世界新药研发的重要贡献。
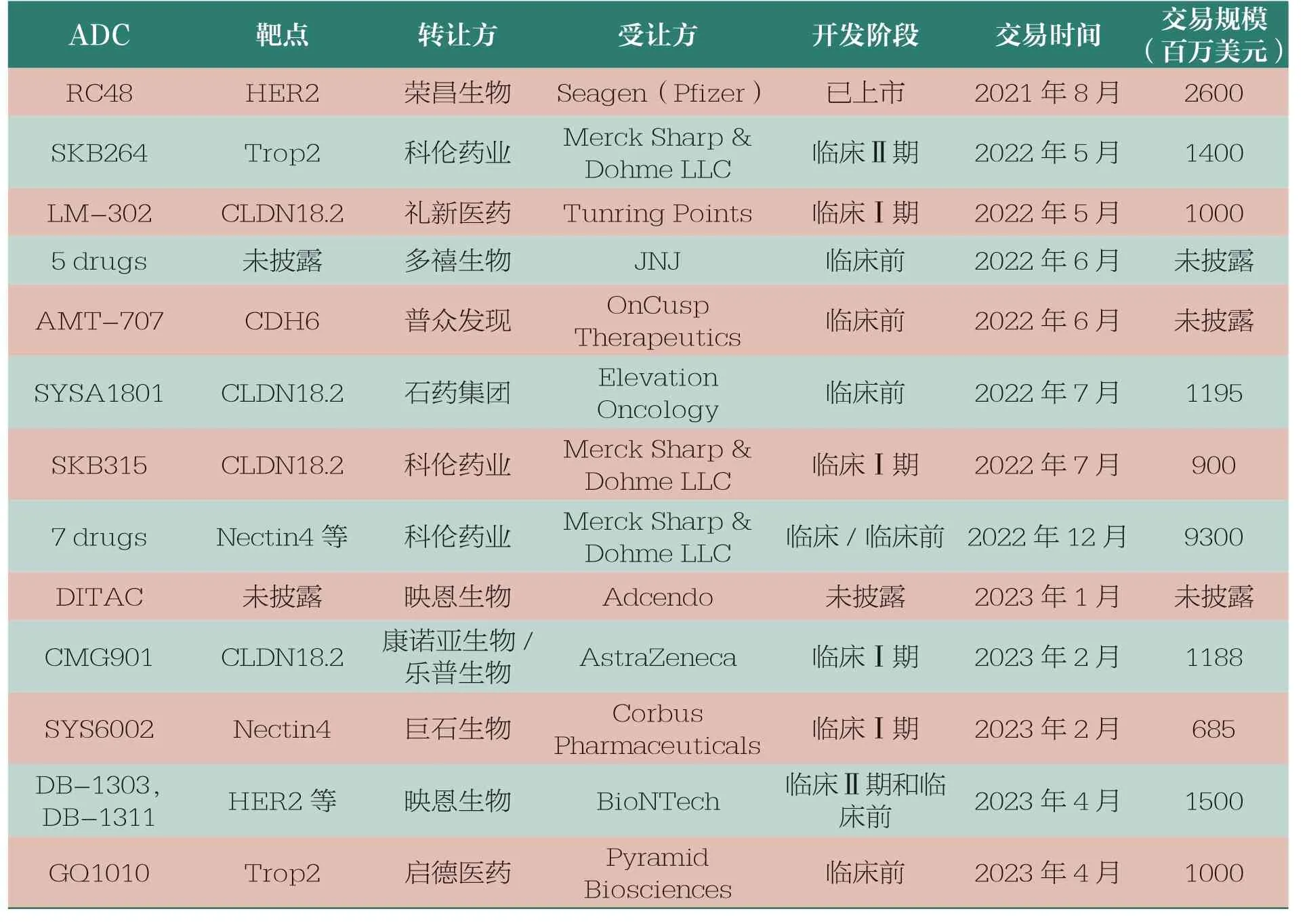
表3 国内药企部分ADC 研发项目国内外转让情况
我国生物医药企业在ADC领域的快速发展是建立在近20年来主要由海归力量衍生出的有强大生命活力的生物医药创新基础之上的。近年来的大规模抗体开发与传统的小分子细胞毒素的结合为国内的创新生物医药企业展示了创新发展方向,资本的涌入也促使药企加入ADC 赛道。表3 中转让项目的同质化情况比较严重,在新靶点及新机制方面仍相对薄弱。另外,使用小分子毒素作为细胞杀伤弹头的选择也较为局限。国内的ADC 开发大多建立在这些常见毒素和细胞杀伤的机制上,在新毒素和新机制应用等方面还需要进一步努力。表4列举了目前部分已获批或开发的ADC 相关毒素弹头。
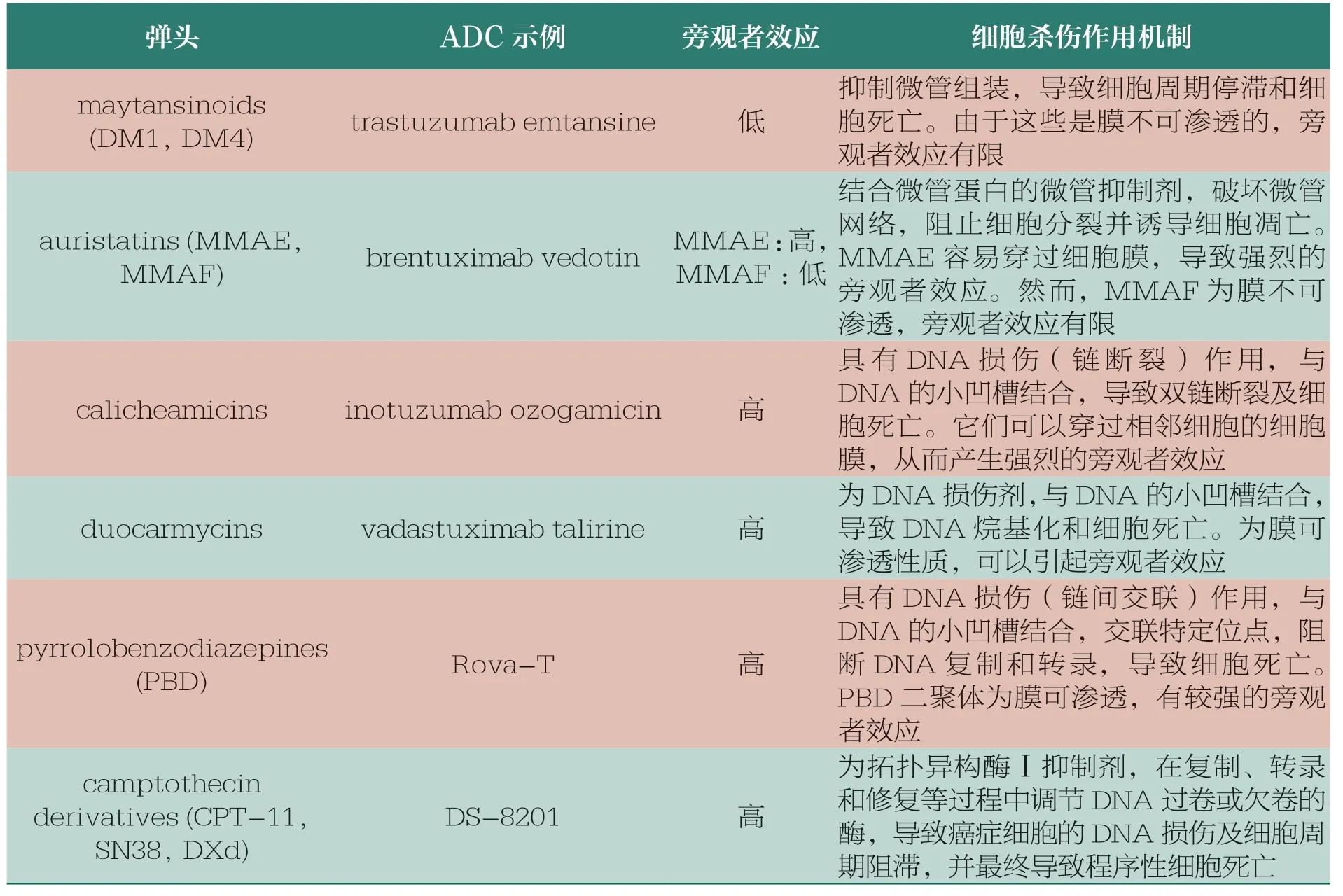
表4 部分ADC 使用的弹头及其细胞杀伤机制
3 ADC 研发的关键因素
自ADC 首次进入临床试验以来,已有数百个ADC 启动了千余项临床试验。在此过程中,多个ADC 的临床试验失败或被从管线中移除。纵观ADC 临床试验失败的原因,安全性和有效性是关键[2]。
3.1 安全性因素
安全性原因主要指ADC 自身的不可耐受的毒性限制了患者用药。其中ADC 的靶向性和连接子毒素的稳定性等对安全性影响较大,无论是靶向非肿瘤细胞还是毒素的非特异性释放都会带来严重的毒性问题。表5 展示了部分因安全性因素被迫终止的ADC。
以靶向CD44v6 的ADC bivatuzumab mertansine 为例,其于2002年启动Ⅰ期临床试验,但随后患者出现了严重甚至危及生命的皮肤脱落不良反应,进而被迫终止临床试验。分析其终止的主要原因在于CD44v6除在肿瘤细胞表达外,还在人体皮肤表面角质层上表达,造成的非瘤靶向结合(off-tumor/ontarget)是致使bivatuzumab mertansine 试验失败的主要原因。ATRC-301 为靶向EphA2的ADC,该药物在非人灵长类动物毒性研究中出现出血等安全隐患,最终被Atreca 放弃。MEDI547 同样为靶向EphA2 的ADC,也是因为非瘤靶向结合致使患者用药后出现了出血、凝血、皮肤和神经系统等方面的反应,最终被迫终止试验。Mylotarg为全球第一个上市的ADC,靶向CD33,用于治疗急性髓性白血病。Mylotarg 曾在2010年因为严重的致命性肝损伤等安全性问题,死亡率远高于对照组而被迫撤市;后来在2017年调整了相应的给药方案,以降低剂量的方式被FDA 批准重新上市。Mylotarg 采用的连接子是含腙键和二硫键的化合物,分析其中间撤市的主要原因在于该连接子不稳定,进而致使Mylotarg 在还未到达靶细胞时,细胞毒素药物卡奇霉素过早释放,引发了致命的毒性,而被迫退市。Trop2 表达于皮肤等正常的上皮细胞,靶向Trop2 的ADC PF-06664178在临床试验阶段出现皮疹等严重的皮肤毒性,被迫终止临床。Trodelvy 同样为靶向Trop2 的ADC,其临床阶段的皮肤毒性要明显低于PF-06664178,已于2020年获批上市。同样靶向Trop2,PF-06664178 搭载的载荷是微管抑制剂auristatin,Trodelvy 搭载的载荷是拓扑异构酶I 抑制剂,因此对于Trop2 这类表达较为广泛的抗原,载荷的选择也是影响ADC 安全性和临床成败的关键。
3.2 有效性因素
有效性方面主要指ADC 的疗效不足,其带来的治疗益处不优于当前的标准治疗。ADC 的药动学(pharmacokinetics,PK)特性差或免疫原性高可导致ADC体内清除迅速,另外ADC 的有效载荷效力不足、有效载荷肿瘤内释放不完全、靶点抗原密度低、ADC 的内化特性差等都是制约ADC 有效性的潜在因素。表6 展示了因有效性因素被迫终止的部分ADC。

表6 因有效性因素被迫终止的部分ADC
以靶向HER2 的SBT6050为例,这是偶联了TLR8 的免疫激动型ADC,意在激活人体免疫系统起到杀伤肿瘤的效果。从其I/Ib 期临床试验的中期数据来看,客观缓解率仅为7%,分析其失败的主要原因在于载荷TLR8的效力不足。Silverback 研发的靶向Nectin4 的SBT6290亦是如此,表明这一策略存在机制缺陷。百奥泰公司开发的BAT8001 和BAT8003 均使用美登素衍生物作为毒素载荷,这两款ADC 相继失败和终止临床的原因可能也是在于美登素衍生物的效力不足。PK 和免疫原性等特性一定程度上决定了ADC 的循环半衰期和体内清除速度,从而也一定程度上影响了ADC 的有效性。MEDI4267(HER2 靶向)在最大耐受剂量下的半衰期很短,极差的PK 使得其在体内被很快清除,进而制约了其临床疗效。SGN-15(FUT3 靶向)和cantuzumab mertansine(MUC1 靶向)等多个ADC也是由于PK较差或免疫原性较高等问题导致临床有效性低而被迫终止。此外,rovalpituzumab tesirine(DLL3 靶向)、depatuxizumab mafodotin(EGFR VIII 靶向)、AGS16F(ENPP3 靶向)以及ifastuzumab vedotin(NaPi2b靶向)等ADC 也因未能证明优于标准治疗而被终止。
3.3 商业化因素
除安全性和有效性之外,商业化因素也左右着ADC 的临床走向。表7 展示了因商业化因素被迫终止的部分ADC,这些均与市场竞争环境及企业的战略调整等原因相关。

表7 因商业化因素被迫终止的部分ADC
4 ADC 的临床毒性
ADC 具有特殊的分子构造,分别由抗体、连接子和毒素弹头组成。在发挥功能时,由具有靶向作用的抗体分子将具有杀伤作用的有效载荷靶向运输到肿瘤部位杀伤肿瘤。与单独的小分子给药相比,ADC 由于靶向性优势可以有效提高药物的治疗效果。目前已上市的ADC 也确实展现出了更优的治疗效果。但是,ADC的毒性依然是不容忽视的问题。如前文所述,目前有大量的ADC由于安全性问题在临床前和临床阶段被终止,并且FDA 已批准上市的12 个ADC 中有10 个被添加了黑框警告。目前的研究表明,ADC 的主要毒性来自毒素部分。Colombo 等[3]将ADC 和小分子单药的剂量进行标准化处理后发现,ADC 的最大耐受量与相应小分子毒素之间并无显著差异。这主要是由于在实际给药时,通常只有1%的ADC 可以达到肿瘤部位,而大部分ADC 仍停留在血液循环中或分布到其他正常组织中。ADC 的非特异性摄取和裂解是其毒性产生的关键。除毒素载荷之外,不同靶点的组织分布和抗体分子的选择也是ADC 产生毒性的重要因素,可以造成药物的非瘤靶向结合等。为了更清晰地对比不同因素在ADC 毒性中的作用,本文分别汇总并分析了偶联MMAE 的ADC 和靶向HER2 的ADC 的不良反应。
4.1 偶联MMAE 的ADC不良反应比较
目前研究认为,ADC 中的小分子毒素是产生机体毒性的主要原因,在临床中现有ADC 所展现出的不良反应大多与这些毒素的表现相似。其中非瘤非靶结合是最关键的因素之一,主要表现为以下几点:①由于连接子稳定性导致的脱靶,即有效载荷在血液循环中的过早释放,释放的有效载荷会进入正常细胞造成细胞杀伤。②由于ADC 疏水性及电荷导致的非特异性内吞,通常疏水性和正电性更高的ADC 会导致更强的非特异性内吞。③ADC分子与免疫细胞表面Fcγ 受体结合导致的内吞,从而出现免疫细胞毒性。以MMAE 为例,表8 汇总了5 个以MMAE 为有效载荷的ADC 和MMAE 分子的不良反应。从表8 可以看出,5 个ADC 均表现出了血液毒性(嗜中性粒细胞减少、血小板减少等)及神经系统毒性等,这些不良反应均与MMAE 的毒性表现相同。

表8 以MMAE 为有效载荷的ADC 不良反应及靶点分布分析
非瘤靶向结合也是ADC 毒性产生的关键因素之一,这主要是由于肿瘤相关靶点的组织分布不同导致的。同样对5 个以MMAE 作为有效载荷、靶向不同靶点的ADC 的不良反应进行对比。从表8 可以看出,除了MMAE 导致的血液/神经毒性以外,这5 种ADC 的毒性反应存在显著差异,这一差异与不同靶点的组织分布存在一定的相关性。例如,enfortumab vedotin、brentuximab vedotin、tisotumab vedotin 和维迪西妥单抗均表现出了胃肠道毒性和皮肤/肌肉毒性。对靶点分布进行调研可以发现,Nectin4、CD30、TF 和HER2 在消化道和皮肤/结缔组织中均具有较高的表达。相对应的,CD79B 在皮肤/结缔组织中不表达、在消化道中低表达。因此,尽管MMAE 具有肠道毒性和皮肤/肌肉毒性,最终靶 向CD79B 的polatuzumab vedotin 也未表现出相关毒性。此 外,enfortumab vedotin 和brentuximab vedotin 的胎儿毒性、tisotumab vedotin 的眼毒性和呼吸系统毒性也均与靶点分布具有相关性。
综上所述,尽管大部分情况下有效载荷本身的毒性决定了ADC 的毒性,但是不同靶点的选择同样也会对ADC 的毒性产生影响。因此,寻找肿瘤分布特异性更高的靶点也是降低ADC毒性的关键解决方案之一。
4.2 靶向HER2 的ADC不良反应比较
除了有效载荷和靶点选择之外,抗体分子自身的性质是否会对ADC 的毒性产生影响?为了回答这一问题,本文对已上市的3 个HER2 靶向的ADC 的不良反应进行了比较(表9)。其中ado-trastuzumab emtansine和fam-trastuzumabderuxtecan 使用的为同一个抗体分子,即trastuzumab,两者分别偶联了DM1 和Dxd 作为有效载荷。维迪西妥单抗则是不同的HER2 靶向抗体,并偶联了MMAE 作为有效载荷。从表9 可以看出,尽管3 个ADC由于HER2 组织分布等因素导致了相似的血液毒性、胃肠道毒性、肝毒性和皮肤/肌肉毒性,但是在呼吸系统毒性和心肌毒性上产生了较大的差异。adotrastuzumab emtansine 和fam-trastuzumab deruxtecan均表现出了间质性肺疾病和心力衰竭等呼吸系统毒性或心肌毒性,这一表现与trastuzumab 单药的表现基本相同。相对应的,尽管HER2 分子在心肌细胞中低表达,并且呼吸系统毒性和心肌毒性均属于MMAE 的毒性反应(表8),但是维迪西妥单抗却并未表现出严重的呼吸系统毒性和心肌毒性,以有效载荷自身的毒性和靶点分布等原因均无法完美解释维迪西妥单抗的这一表现。目前主流观点认为,靶向HER2 的抗体药物(trastuzumab,pertuzumab,ado-trastuzumab emtansine 和fam-trastuzumab deruxtecan)的心肌毒性与HER2 信号传导通路被抑制有关,那么维迪西妥单抗表现出的心肌毒性差异则可能与其阻断功能相关,对此需要对这几种抗体的生物学功能进行深入研究和对比。

表9 以HER2 为靶点的3 种ADC 的不良反应分析
综上分析,ADC 毒性是抗体及连接子毒素的综合表现,其中小分子毒素的最大耐受量决定了ADC 的体内应用情况。对非瘤非靶结合及非瘤靶向结合等的综合分析,从平衡小分子毒素与抗体的结构组成以及抗体优化方面共同努力,对于减少ADC 的不良反应将有所帮助。
5 ADC 的内吞与药效
理想情况下,ADC 在进入靶细胞之前在血液循环中应保持稳定,但这一生物大分子不能通过被动运输的方式透过细胞膜进入细胞,需要抗体与细胞膜上的靶点蛋白结合后再被内吞进入细胞。一般来说抗体或ADC 的内吞过程可以分为以下3 个阶段:①抗体与细胞膜表面靶点抗原结合;②细胞膜内陷形成内吞囊泡;③内吞囊泡从细胞膜脱离进入细胞质(图1)。由于存在多种内吞途径,而且各途径之间存在相互影响,因此内吞过程是高度灵活且复杂的,这也导致了目前对于ADC 内吞作用与药效之间的研究并不透彻[4]。
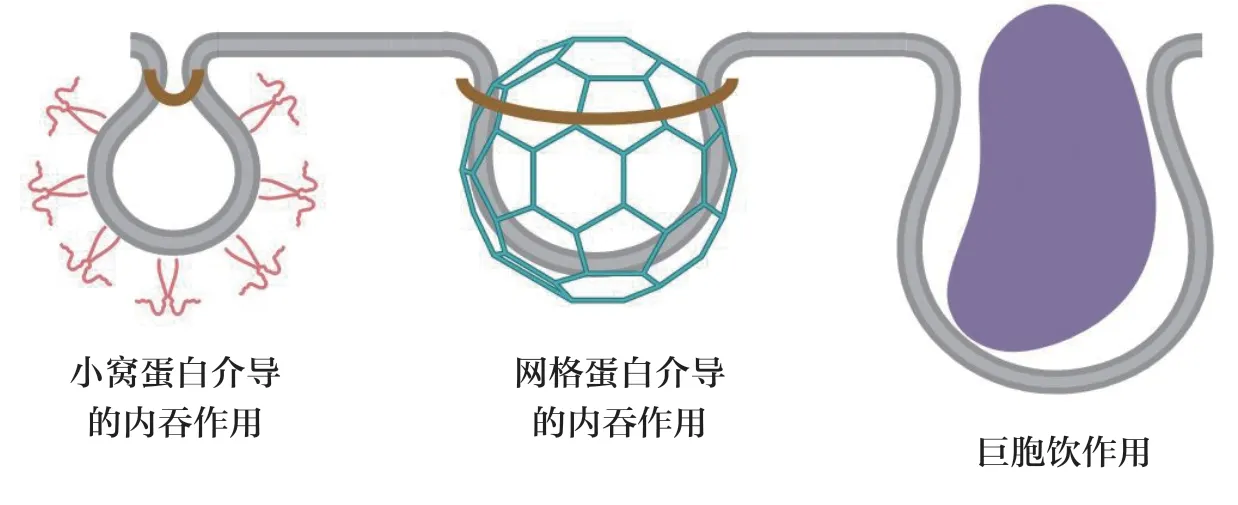
图1 ADC 常见的3 种内吞途径
5.1 网格蛋白介导的内吞作用
ADC 与细胞膜表面的靶点蛋白结合形成复合物,网格蛋白及一系列相关作用分子使细胞膜内陷形成包被小窝,包被小窝在逐渐增大的同时被肌动蛋白拉入细胞内,直至从细胞膜脱离下来形成网格蛋白包被囊泡,然后通过核内体-溶酶体途径进行胞内代谢。多种ADC 被证实与网格蛋白共定位,并且经网格蛋白抑制剂处理后,ADC 内吞速率明显被抑制,因此该内吞作用被证实为ADC 内吞的主要形式[5-6]。网格蛋白由1 条重链和1 条轻链组成,3 个网格蛋白的重链和轻链形成1个“三棱体”,并与其他“三棱体”相互作用在包被小窝周围形成一个多边形晶格。囊泡的形成需要50 多种胞质内蛋白有序的参入。
5.2 小窝蛋白介导的内吞作用
小窝是位于细胞表面的一种瓶状的细胞质膜凹陷结构,以较高水平的胆固醇和鞘糖脂为特征,存在于大多数细胞类型中。部分ADC 也可以通过该途径进入细胞,并转运至内质网和高尔基体内,但这两种细胞器缺乏使ADC裂解的酶和环境因素,因此该内吞途径可能与ADC 发挥药效无关[7]。尽管小窝蛋白介导的内吞与网格蛋白介导的内吞都能使细胞膜发生凹陷,但小窝蛋白形成的瓶状凹陷在内吞过程中的大小保持不变,而且在各细胞株中的密度存在很大差异[8]。小窝蛋白1(Cav-1)是小窝的主要结构蛋白,除参与内吞外,还参与多种细胞活动,如细胞黏附、信号转导等。有研究表明,小窝蛋白与耐药机制有关,例如Cav-1 被发现可以负向调节HER2 膜上表达影响抗HER2 药物的疗效[9-10]。
研究表明,Cav-1 与HER2共定位于细胞膜凹陷结构,影响HER2 在癌细胞膜上的表达和稳定性。HER2 的表达与Cav-1呈负相关,Cav-1 高表达的细胞HER2 膜表达低,且倾向于胞内积聚(图2),在体内肿瘤模型如NCI-N87 异种移植瘤中可以发现这样的负相关表达区域。在异质性较高的患者肿瘤组织中,Cav-1 高表达造成肿瘤细胞膜HER2 低表达,会影响肿瘤组织对HER2 药物的临床疗效。HER2 和Cav-1 的逆 向调节及对抗HER2 药物的影响已在动物实验中证明,抗脂类代谢药物lovastatin 通过抑制Cav-1表达,可以提高肿瘤细胞HER2的膜表达,并进而增强T-DM1的抗肿瘤药效[9-10]。以上发现具有一定的临床指导意义,或许可以通过与lovastatin 等胆固醇类药物联用治疗HER2 低表达或T-DM1 耐药患者。

图2 HER2 与Cav-1 在肿瘤组织共表达模式图
5.3 巨胞饮作用
部分ADC 也可以通过巨胞饮作用进入细胞内,该作用是细胞对胞外大分子物质的一种非特异性的内吞作用[11]。例如,Jedema 等[12]报道治疗急性髓性白血病的ADC gemtuzumab ozogamicin(CD33 靶 向)存在2 种内吞途径,在低浓度时通过与细胞表面的CD33 结合启动网格蛋白介导的内吞,而在高浓度时通过巨胞饮作用进入CD33阴性细胞,这2 种内吞途径都能释放有效载荷阻断DNA 复制,影响细胞周期,导致细胞死亡。但巨胞饮作用作为一种非特异性的内吞作用,往往与不良反应相关,例如,临床中观察到的gemtuzumab ozogamicin 肝毒性可能与肝脏中存在大量具有巨胞饮作用细胞有关。
ADC 的内吞与胞内转运对疗效及毒性有着显著影响,提高内吞速率、加快ADC 向核内体-溶酶体聚集有可能提高ADC 对肿瘤细胞的杀伤并减少毒性。由于ADC 开发周期长、耗费大,为了提高ADC 开发的成功率,需要谨慎选择靶点、抗体结合表位及亲和力、连接子的类型、有效载荷,以上各项都有可能影响ADC 的内吞速率及胞内转运。在靶点蛋白的膜表达方面,分析抗原低表达机制对提高药效和解决耐药等有重要意义。结合其他药物可能导致的肿瘤抗原膜表达变化情况,对不同患者群体开展深入分层研究,可以进一步提高对患者及ADC 治疗的认识,也为已上市的ADC 与其他药物联用增强疗效提供临床用药创新策略。
6 结合位点屏障作用对ADC 的影响
ADC 作为一种靶向杀伤肿瘤的抗体药物已越来越受到关注,但是在临床实践中,ADC的疗效受到多种因素的制约。例如,在实体肿瘤的治疗中,ADC面临的另一个难题就是药物在肿瘤组织渗透中的结合位点屏障(binding-site barrier,BSB)。当ADC 从血管进入实体瘤组织中,高亲和力的抗体通常会紧密结合在血管周围的实体瘤细胞,难以向肿瘤内部渗透,导致ADC仅对血管周围表层细胞进行杀伤而难以进入实体瘤核心(图3),从而影响药效[13-14]。从图3 可以看出,抗体的亲和力及靶点的分布情况均会影响ADC 的肿瘤组织穿透,因此在选择ADC 抗体时需要结合靶点的具体情况对抗体结合的亲和动力学作择优比较,才能保证ADC 有较好的肿瘤组织穿透能力和药效。理论上,增大给药剂量在一定程度上能解决渗透性差的问题,但对于ADC来说,药物毒性限制了给药剂量,因此增大剂量的方法不适用。这里主要介绍2 种增强ADC 肿瘤组织渗透的方法:ADC 与其对应的裸抗体联合给药[14];ADC 与即时竞争抑制性抗体联合给药[15]。

图3 影响ADC 穿透的结合位点屏障现象
6.1 ADC 与其对应的裸抗体联合给药
该方法采用毒素偶联的抗体ADC 和未偶联的裸抗体联合给药,混合物中裸抗体的含量为ADC 的5~10 倍。高浓度的裸抗体分子竞争结合实体瘤血管表层细胞较多,促使ADC 进一步向实体瘤内部渗透。如图4 所示,靠近血管的肿瘤细胞上的靶点被ADC 及裸抗体分别结合占位,其中裸抗体结合优势较高,促使血管中ADC 渗透进入距血管距离更远的肿瘤组织,实现与肿瘤内部靶细胞的结合。该方法能在不增加ADC 给药剂量和药物毒性的前提下,有效增加ADC 渗透能力以及最终药效。目前,该方法在众多抗体药物、针对不同靶点的实体瘤临床前实验中得到了验证。例如,抗表皮生长因子受体(EGFR)抗体panitumumab 偶联荧光分子IRDye800CW模拟的ADC,可以通过检测荧光信号确定panitumumab-IRDye800CW的肿瘤组织分布情况。结果发现,panitumumab-IRDye800CW单药组荧光信号主要分布在实体瘤血管表层,而panitumumab 和panitumumab-IRDye800CW混合组荧光信号分布更深入实体瘤内部[14]。在T-DM1 和trastuzumab 联用中也发现类似的实验结果[16]。在此种联合给药策略下,ADC/裸抗体混合物的渗透能力与ADC/裸抗体含量比例相关。在ADC 含量不变的条件下以及一定的剂量浓度范围内,裸抗体的含量越高,ADC 的渗透能力越强[16-18]。虽然众多临床前研究表明该方法有助于增加ADC 的渗透性以及药效,但该方法的临床有效性还需要进一步证明,仍存在一系列的问题需要解决,诸如ADC/裸抗体的混合比例、靶细胞抗原丰度、裸抗体本身的不良反应等。
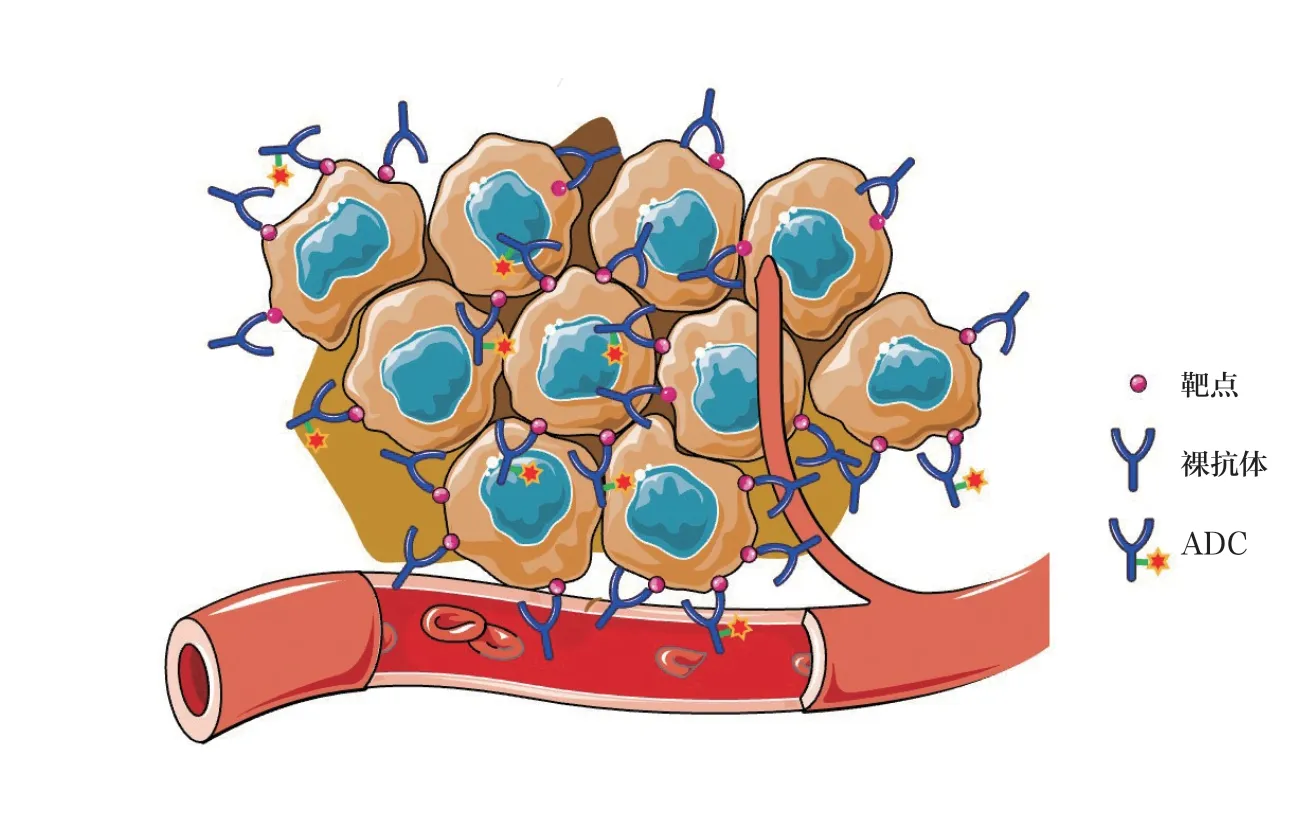
图4 ADC 与裸抗体联合给药增强ADC 渗透
6.2 ADC 与即时竞争抑制性抗体联合给药
该方法的主要思路是开发一个结合抗ADC 的抗体结合域的抗体(anti-idiotypic antibody)。将该抗体与ADC 混合使用,抗ADC 抗体与ADC 的抗体结合域结合后,降低ADC 与抗原的结合,避免ADC 在实体瘤血管表面富集、结合和内吞,促进复合物向实体瘤内部渗透。当进入实体瘤内部后,抗ADC 抗体脱落,解除对ADC 抗体结合域的封闭并快速降解,进而释放出可以结合靶点抗原的ADC,并发挥其肿瘤杀伤作用。例如,Alvarez-Reuda等[19]开发出抗trastuzumab 的羊驼单域抗体1HE。该抗体与trastuzumab 联合给药时能有效降低trastuzumab 与HER2的结合,同时还具备1HE半衰期短(1.2小时)但1HEtrastuzumab 复合物半衰期长(56 小时)的特性,有效地保证该复合物在血液、实体瘤环境中的稳定性以及渗透实体瘤内部1HE 解离后的快速降解。1HET-DM1 复合物在小鼠SK-OV3和NCI-N87 移植瘤模型中的肿瘤渗透能力以及抗肿瘤药效均强于T-DM1 单独给药[15]。
虽然上述方法在体外实验和小鼠体内实验均展现出良好的增强ADC 渗透效果和提高ADC药效的作用,但相关人体试验还未完成。上述联合用药以期提高ADC 的临床疗效还有待于进一步的临床试验来验证。如何制定有效的临床开发策略还需要更多的临床前研究提供支持,同时需要谨慎的临床方案设计,并持续不断地优化方案来保证临床试验的成功。总的来说,开发多元化策略提高ADC 肿瘤组织穿透、提高药效、降低ADC 毒性、建立有效临床试验和治疗方案是ADC开发成功的重要保障,如何让ADC 有更好的肿瘤组织穿透和杀伤是ADC 设计及临床应用中需要不断探索的。
7 ADC 的延伸——抗体介导的RDC
放射性RDC 是含有放射性核素供医学诊断和治疗用的一类特殊药物。与ADC 类似,在结构上RDC 主要由配体、连接子、螯合物和放射性同位素构成。RDC 与ADC 的最大差异是药物载荷;RDC 不再是小分子毒素,而是放射性核素。使用不同的医用核素,可以开展显像或治疗的不同功能,部分核素兼备2 种能力。诊断用核素药物用于获得体内靶器官或病变组织的影像或功能参数,进行疾病诊断;治疗用核素药物将细胞毒性水平的放射性核素输送到病变部位,利用放射性同位素辐射产生局部电离辐射生物效应,对病变细胞或组织产生杀伤作用。
7.1 RDC 开发进展
放射性药物的开发已有较长历史,早期获批的放射性药物大多是非靶向性药物,利用放射性同位素本身的体内富集特点对患者进行治疗,同时会杀伤正常细胞,不良反应较大。能够实现对目标病灶进行靶向性杀伤的靶向放射性药物研究,近几年也取得了较好的进展(表10)。其中,Lutathera、Pluvicto 等靶向放射性药物的获批,极大地推动了放射性药物精准靶向发展的步伐。
目前在研药物靶点除了对标SSTR 和PSMA,还有实体瘤常见靶点如PD-L1、HER2(图5),其中靶向PD-L1 的RDC让人非常感兴趣。众所周知,放疗可以诱导免疫反应,促进免疫细胞进入肿瘤,但放疗后肿瘤可能出现PD-L1 上调而削弱抗肿瘤免疫,因此靶向PD-L1 的RDC 或许能使免疫治疗不敏感的“冷”肿瘤变“热”,前景让人期待。
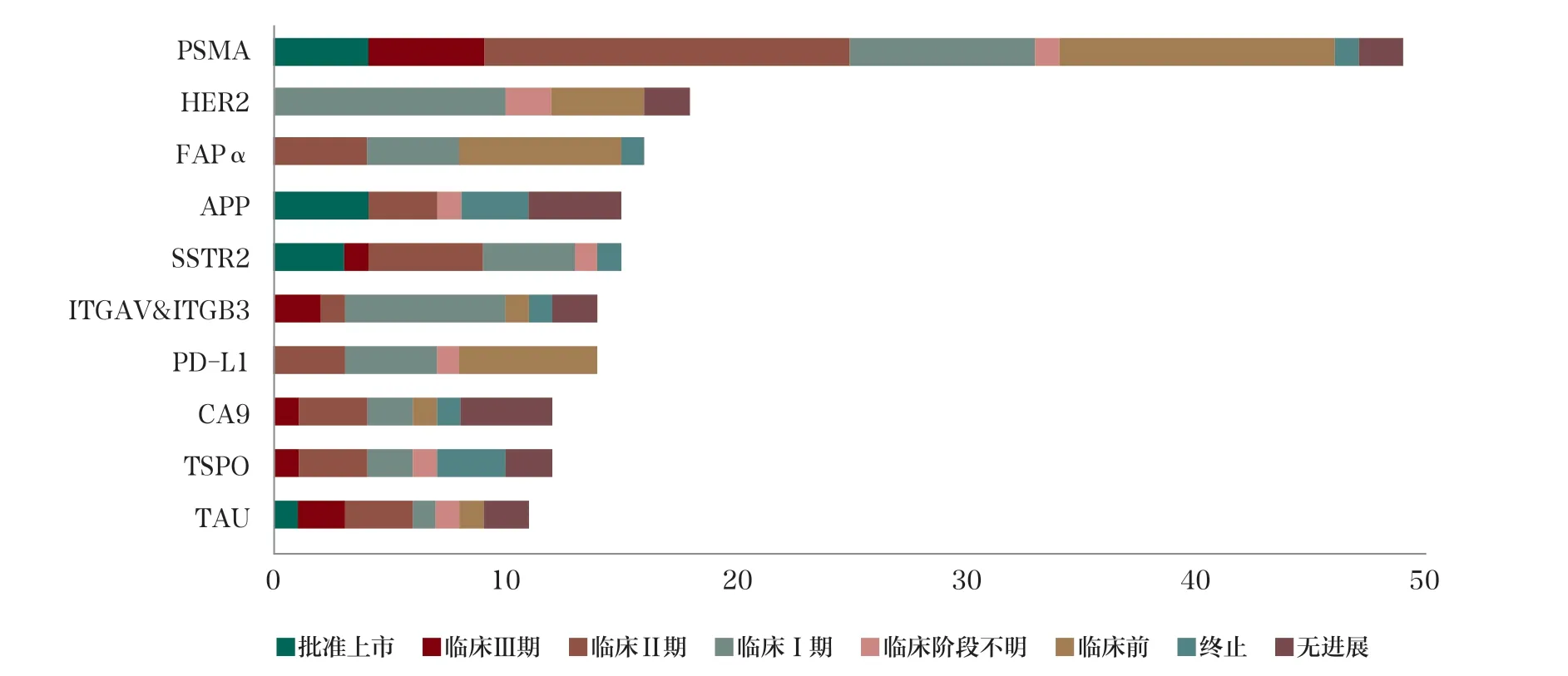
图5 全球放射性产品及体内诊断产品靶点格局TOP10
7.2 RDC 的独特优势
RDC 存在很多ADC 无法比拟的优势,近几年成为肿瘤靶向治疗领域的一颗“新星”,不断有传统药企和创新药企进入。
首先,RDC 是临床实操中唯一能够实现诊疗一体化的药物。RDC 通过构建适当大小可以满足组织穿透的靶向分子和装载半衰期较短的同位素,药物迅速从血液进入靶组织,实现核素与原位肿瘤或继发肿瘤相结合,在极短半衰期内给出信号,并通过分子影像技术得出全面医学影像结果。其特点在于可以只更换核素部分,相关的靶向分子和连接子都保持相似的情况下,就能够形成诊疗一体化的产品,涵盖诊断、分级与分期、治疗、疗效监测及预后判断等过程(表11)。例如,Pluvicto 可以先将诊断性同位素标记在靶向分子和连接子构成的前体上面,然后用PETCT 进行扫描,进而确定患者病灶的位置,以及其目标靶点的表达情况;接下来就可以用治疗性的放射性核素连接成新的治疗性RDC 进行治疗。治疗后还可以再次用诊断性同位素标记的诊断性RDC 进行检测,来观察它的疗效,整个诊断治疗的流程是精准且一体化的。梳理当前国内外RDC 开发管线,诊疗一体化已经成为开发趋势,国外RDC 研发的领先公司,如ITM、Telix Pharmaceuticals、Clarity 等,均已在产品线布局中采用这一策略。
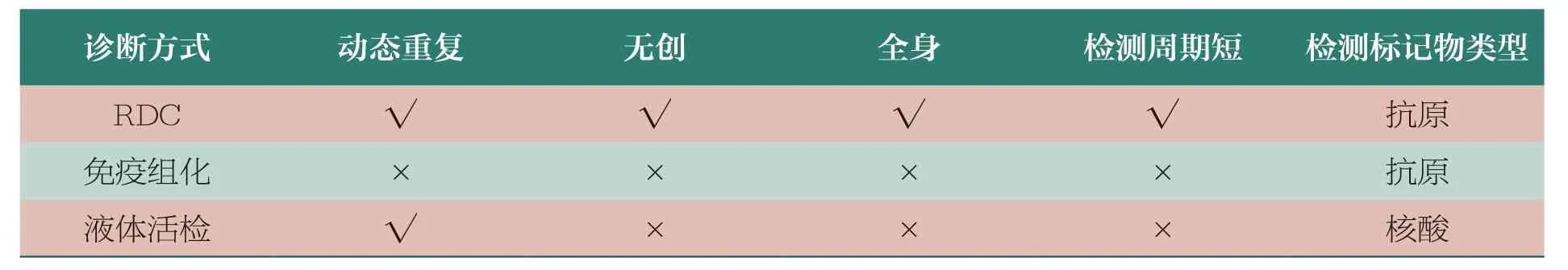
表11 RDC 体内诊断与其他诊断方法对比
其次,由于RDC 作用机制不同,决定了它的治疗优势。主要是基于核素的物理杀伤作用直接或者间接断裂肿瘤细胞DNA,达到杀伤肿瘤的目的。RDC 只需要在目标细胞附近有一定的滞留时间,达到一定剂量的电离辐射就可以杀灭肿瘤细胞,而不需要内吞进肿瘤细胞内。因此,靶向载体的选择范围更为广泛,小分子、多肽或者抗体药物都可以成为RDC 的靶向载体。治疗肿瘤时,RDC 除了直接损伤靶肿瘤细胞外,还具有“交叉火力/旁杀效应”,因此在扩散性肿瘤中具有应用潜力。由于它的杀伤机制跟其他药物不同,所以即便在其他药物无效或者耐药的时候,RDC还是可以发挥治疗作用。例如,已获批的Pluvicto 能够使末线转移性去势抵抗性前列腺癌患者总生存期延长4 个月[20],成为前列腺癌现有治疗方案的有力补充。
7.3 核素标记助力ADC开发
在ADC 早期发现阶段,采用放射性核素标记技术,可以清楚地跟踪候选ADC 分子在体内的PK 行为。对比不同标记位置、不同偶联方式的体内行为差异,有助于尽早筛选出符合预期的ADC 分子。有研究发现,ADC连接子的化学性质对其在临床前动物模型中的吸收、分布、代谢和排泄(ADME)特性有显著影响,特别是对血浆PK 及肿瘤和肝脏组织中观察到的分解代谢物[21]。还有研究证实,ADC 抗体偶联比 值(drug-antibody ratio,DAR)也会影响其在体内的分布和代谢,从而影响药物抗肿瘤效果[22-23]。例如,美登素类细胞毒素ADC 在小鼠体内的PK 分析表明,DAR 平均值在6 以下时,具有较好的PK 清除速率;DAR平均值在9 以上时,清除很快。组织分布结果显示,DAR 值较低的ADC 组织分布特征与裸抗体相似,而DAR 值超过9 的ADC组织分布和裸抗体有明显差异:给药后小分子化合物在全血中的浓度迅速下降,同时很快积聚在肝脏,随后经肝脏快速排出体外,导致体内暴露量降低,对应药效学表现不佳[23]。
在ADC 开发中,非临床研究过程中组织分布、体内代谢和排泄的研究存在诸多挑战,使用放射性核素标记ADC 进行自显影,不仅可以了解整个组织的分布,而且可以区分组织内各区域的浓度差异,再分区域定量,尤其对于评估ADC 在肿瘤组织的分布深度与药效之间的量效关系具有传统方法不可比拟的优势。目前已上市的ADC,如Adcetris、Padcev、Kadcyla、Polivy、Besponsa、Enhertu、Zynlonta、Tivdak 在临床前阶段均采用了低能量放射性核素(14C 或3H)标记小分子化合物,完成了组织分布、物质平衡、血浆蛋白结合率、代谢产物鉴定等实 验。其 中Kadcyla、Polivy、Enhertu、Tivdak 同时采用了放射性核素(125I、111In、3H、89Zr)标记抗体,对比仅标记抗体或小分子化合物后呈现出组织分布差异。例如,Enhertu 使用不同放射性核素分别标记抗体和小分子毒素,通过2 种动物的实验结果,完整阐述ADC 在体内的PK 过程:Enhertu 注射进入体内后,跟随抗体驱动分布至全身各处,全血中浓度最高,并不在正常组织中滞留。在体内释放出的Dxd基本不代谢,绝大部分以原型的形式经胆汁流入肠腔,最终从粪便排出体外[24]。显而易见,RDC技术对ADC 的成药性及优化工作有重要意义。
ADC 开发是一个复杂、漫长、昂贵且容易出错的过程。在过去的几十年里,临床开发的复杂性和成本呈指数级增长,而成功率却没有相应的提高。美国FDA 提出了“探索性研究用新药(exploratory investigational new drug,eIND)”的概念,并于2006年发布了《探索性研究用新药研究指南》(Exploratory IND Studies),以eIND 进行的临床试验又称“0 期临床试验”。0期临床允许对结构相似的一组不超过5 个化合物或剂型进行“微剂量”研究,同时获得人体PK数据,以及采用各种影像学研究手段获得人体组织分布情况,便于早期从一组候选分子中确定最有研发价值的一个先导化合物进行I 期临床试验及后续的研发。放射性核素标记和显影技术在药物0 期临床中发挥着重要的作用。据报道,在116 项已发表的0 期临床研究中,45%使用LC-MS/MS,29% 使 用AMS,23%使用PET 技术[25]。
RDC 的快速发展,扩充了肿瘤标准治疗失败患者的治疗方案,为患者带来更多希望。同时核素标记RDC 的开拓也为ADC 的开发起到了积极的促进作用,可以预期ADC 及RDC 的相互支持对靶向生物药的发展有战略指导意义。当然在RDC 开发中,由于监管状态待完善,核素种类和资源的相对短缺,技术开发的瓶颈、人才短缺等问题,存在不少发展局限性,但也提示RDC 开发在我国有较大提升空间。近年来,RDC 开发中的技术问题,例如高亲和力的小分子多肽筛选困难、小分子RDC 在体内代谢过快、肿瘤部位滞留时间有限而影响疗效、经验证的RDC 靶标相对有限等,都可以从ADC 开发的成功经验中得到更多的资源和启发。
从对靶向生物药ADC 多方面的分析来看,目前此类药物的改进、治疗方案的优化等都有较大提高空间,标示着这一创新药物领域的强大生命力。本文中提到的抗体优选、毒素和连接子创新、ADC 结构优化、肿瘤组织穿透、低靶点表达和耐药、联合治疗策略等问题还需要行业的不懈努力。RDC 更是为ADC 的延伸提供了相互支持的重要力量,结合RDC 在影像技术中强大功能,在早期临床开发中的0 期临床试验创新支持,相信会对ADC 和RDC 靶向生物药的发展起到巨大推动作用,也为靶向生物药的发展和应用开拓更广阔的空间。
