Rh催化C-C键选择性活化反应的密度泛函理论研究进展
2023-02-23李江平
李江平
(云南师范大学,云南 昆明 650500)
C—C键普遍存在于有机分子中,催化C—C键活化可有效改变复杂有机分子的合成方式,特别是从C—C键选择性活化开始,随后将各种不饱和键插入特定的C—C键,对于获得特定的骨架非常有效[1-4]。但因C—C键的动力学惰性和热力学稳定性使得活化C—C键具有很大挑战。值得高兴的是,近年来,在过渡金属催化剂的存在下,可以通过氧化加成、β—C消除和烯丙基化等方式使C—C键活化[5-13]。铑是在催化C—C键选择性活化中起到重要作用的一类过渡金属,它不仅能使张力环分子的C—C键断裂,同时也能辅助导向基团使C—C键选择性活化[14-16]。关于过渡金属催化C-H键活化的理论研究成果不断被总结[17-19],但针对过渡金属催化C—C键活化特别是Rh催化的综述尚未见报道,因此本文对近些年来Rh催化C—C键选择性活化反应的DFT研究作出了综述,以四元环以及直链上的C—C键选择性活化为主线,重点对C—C键活化步骤的机理和选择性进行阐述。
1 Rh催化四元环上的C—C键选择性活化反应
四元环分子存在着巨大的环张力,开环消除环应变可以为C—C键活化提供强大的热力学驱动力,因此,活化四元环上的C—C键是构建复杂分子强有力的手段。
2012年,Xu课题组[20]报道了铑催化苯并环丁烯酮的区域选择性碳酰化反应。为近一步探究区域选择性的来源,2015年,Lu等人[21]对苯并环丁烯酮衍生物发生C—C键活化时是C1—C2键断裂还是C1—C4键断裂进行了详细的DFT研究(如图1所示),计算结果显示C1—C4键通过过渡态TS2-3发生活化时仅需越过 18.6 kcal/mol 的能垒,而通过过渡态TS2-4发生C1—C2键活化则需要高达 39.5 kcal/mol 的能量,此外C1—C4键活化路径得到的中间体INT3比C1—C2键活化得到的中间体INT4稳定很多(-17.2vs1.3 kcal/mol),过渡态TS2-3较TS2-4在热力学上更稳定以及此条路径在动力学上更为可行,由此可推测活化C1—C4键是该反应的较优选择。作者推测这可能与烷氧基离催化剂上的大体积配体的距离有关。

图1 铑催化苯并环丁烯酮C—C键选择性活化 步骤的势能剖面图(能量:kcal/mol)
值得注意的是,2019年,Cheng课题组[22]对Xu等人[20]的实验提出了不同的机理,作者认为铑与环丁酮氧络合后可以选择性活化烷氧基上的双键而不活化环丁酮上的C—C键。计算结果显示(如图2),无论是R(虚线)还是S(实线)反应的C—C键活化均可一步完成,且S反应的C—C键活化(TS5-7:ΔG=8.3 kcal/mol)在能量上比上述Lu等人[21]的多步机理更占优势。

图2 铑催化苯并环丁烯酮C—C键选择性活化 步骤的势能剖面图(能量:kcal/mol)
2017年,Qin课题组[23]报道了2-烷基苯并环丁酮扩环反应的DFT研究,计算结果显示(如图3)该反应主要通过氧化加成,β—H消除,氢化和C—C还原消除等步骤发生。当苯环上不存在取代基时,更倾向于C1—C2键活化开环,仅需要跨越 23.5 kcal/mol 的自由能垒(TS14-15)。
2019年,Deng课题组[24]报道了铑催化环丁酮C—C键活化合成5,6-稠合双环和C2-取代环丁酮的反应,并利用DFT对选择性的来源进行了研究。计算结果显示(如图4)环丁酮C—C键活化存在三条路径,其中氮甲苯磺酰基氧与铑中心的弱配位扮演着重要角色,弱配位能有效稳定关键结构。因此最有利的C—C键活化通过过渡态TS17-18(ΔG=22.0 kcal/mol)发生,取代较多的C—C键活化并得到中间体INT18。经过渡态TS19(ΔG=27.1 kcal/mol)和TS20(ΔG=29.1 kcal/mol)的C—C键活化因缺乏配位需要跨越更高的能垒,与实验中观察到的高选择性一致。在此文中最少取代的C—C键的活化在动力学上是最不利的,即位阻较小的C—C键更不容易被活化。
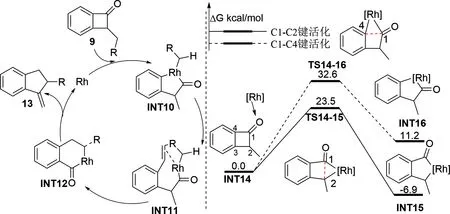
图3 铑催化2-烷基苯并环丁酮扩环反应的机理以及部分势能剖面图(能量:kcal/mol)
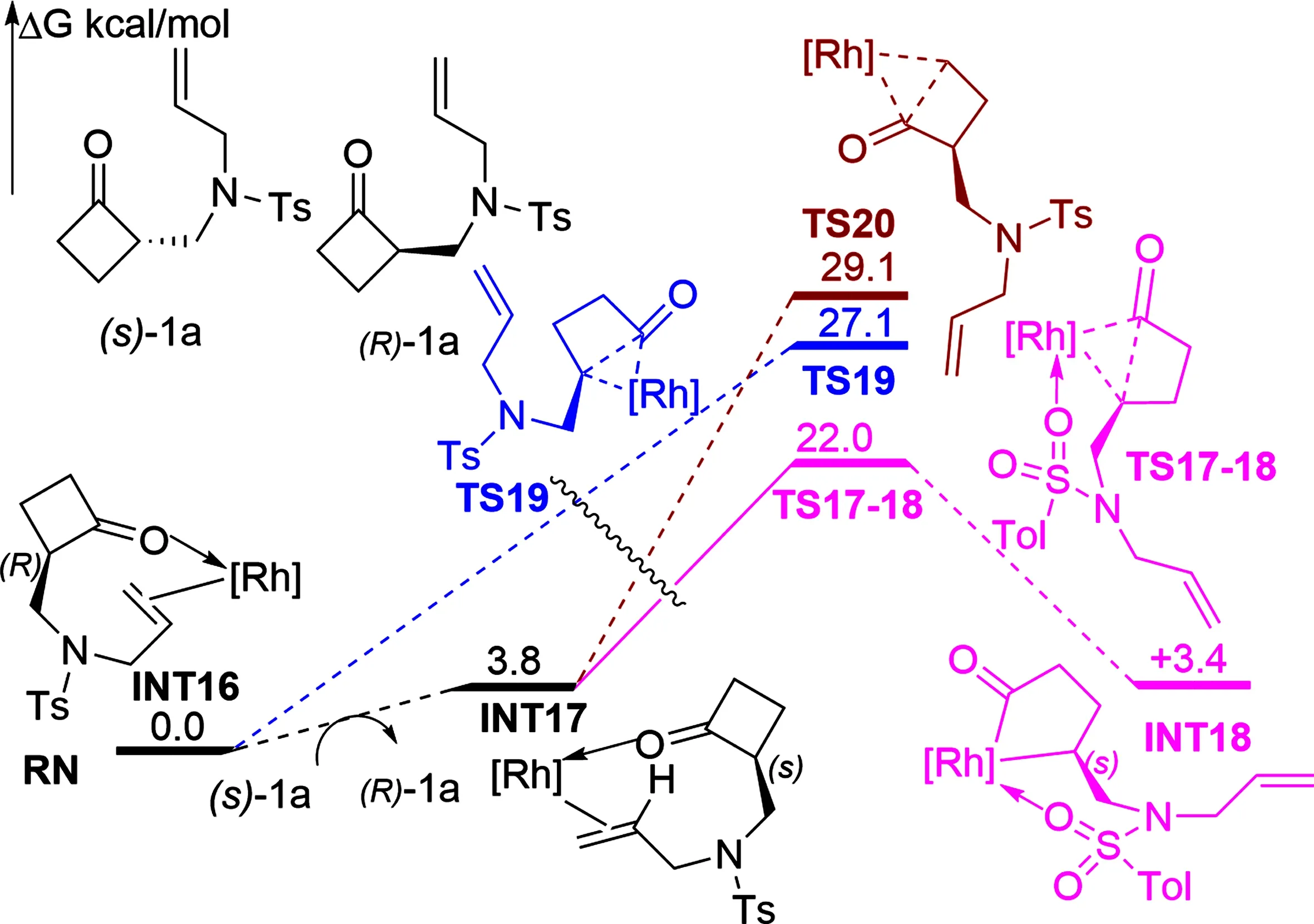
图4 铑催化环丁酮C—C键选择性活化步骤的势能剖面图(能量:kcal/mol)
配体会显著影响C—C键活化的能垒。基于此,2020年,Ham等人[25]尝试使用铑催化α-羟基β-内酰胺的C—C键活化来合成氮稠杂环(如图5)。令人出乎意料的是,添加配体不仅能有效降低C—C键的活化能垒,还能使其出现选择性。经DFT计算得出此反应在配体Xantphos的协助下,远端C—C键经过渡态TS24-26活化需 12.8 kcal/mol 的能量,而近端C—C键活化(通过过渡态TS25-27)需 18.6 kcal/mol 的能量,结合连接到铑中心的酰胺酰基和烷基之间的强度差异导致INT26和INT27之间的结构差异,使得INT26更稳定(两者之间能量差 6.4 kcal/mol),所以观察到的远端C—C键断裂路径是有利的,这与实验结论一致。
2020年,Zhang等人[26]在之前研究的基础上对环丁酮的开环反应做了一步的理论研究。计算结果显示(如图6),对于C2取代的环丁酮可经过渡态TS29-30(ΔG=14.3 kcal/mol)发生C1—C2键活化,或经过渡态TS29-31(ΔG=7.2 kcal/mol)发生C1—C4键活化,后者较于前者低 7.1 kcalmol-1,则远端的C—C键断裂更有优势,与上述结论一致,作者认为相互作用及畸变能导致了这种选择性,此结果与实验观察一致。
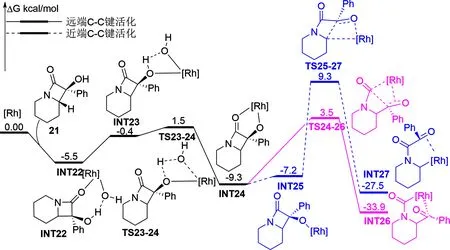
图5 铑催化α-羟基β-内酰胺C—C键选择性活化步骤的势能剖面图(能量:kcal/mol)
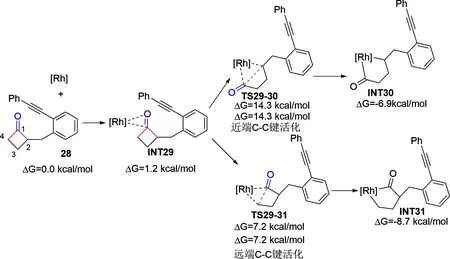
图6 铑催化环丁酮C—C键选择性活化步骤的势能剖面图(能量:kcal/mol)
2 Rh催化直链上的C—C键选择性活化反应
链状化合物因不具有环张力而较为稳定,因此C—C键活化大多只存在具有环应变或使用导向基团的体系。
鉴于此,2018年,Kong小组[27]对铑催化1,4-二苯基丁基-3-炔-1,2-二酮羰基化过程中的反应选择性开展了理论研究。计算结果显示使用Xantphos作为配体时,反应经历了两次氧化加成,在第一次氧化加成中,C—C键活化存在三条路径(如图10),当铑催化底物的羰基和羰基之间的C—C键通过过渡态TS33-34活化时需要跨越的能垒为 43.1 kcal/mol。而经历过渡态TS35-36(或TS37-38)时底物的芳基和羰基之间的C—C键(或底物炔基和羰基之间的C—C键)发生活化,此时需要越过的能垒为30.0 (或27.0) kcal/mol。根据能垒,通过过渡态TS37-38发生C—C键活化是最为有利的。在第二次氧化加成中,芳基C与羰基C(TS39-40)之间的活化能垒较羰基C与炔基C(TS39-41)间的活化能垒高 21.0 kcal/mol。因此两次氧化加成均是底物炔基和羰基之间的C—C键活化最为有利,这可能与C—C键之间的极性大小有关。
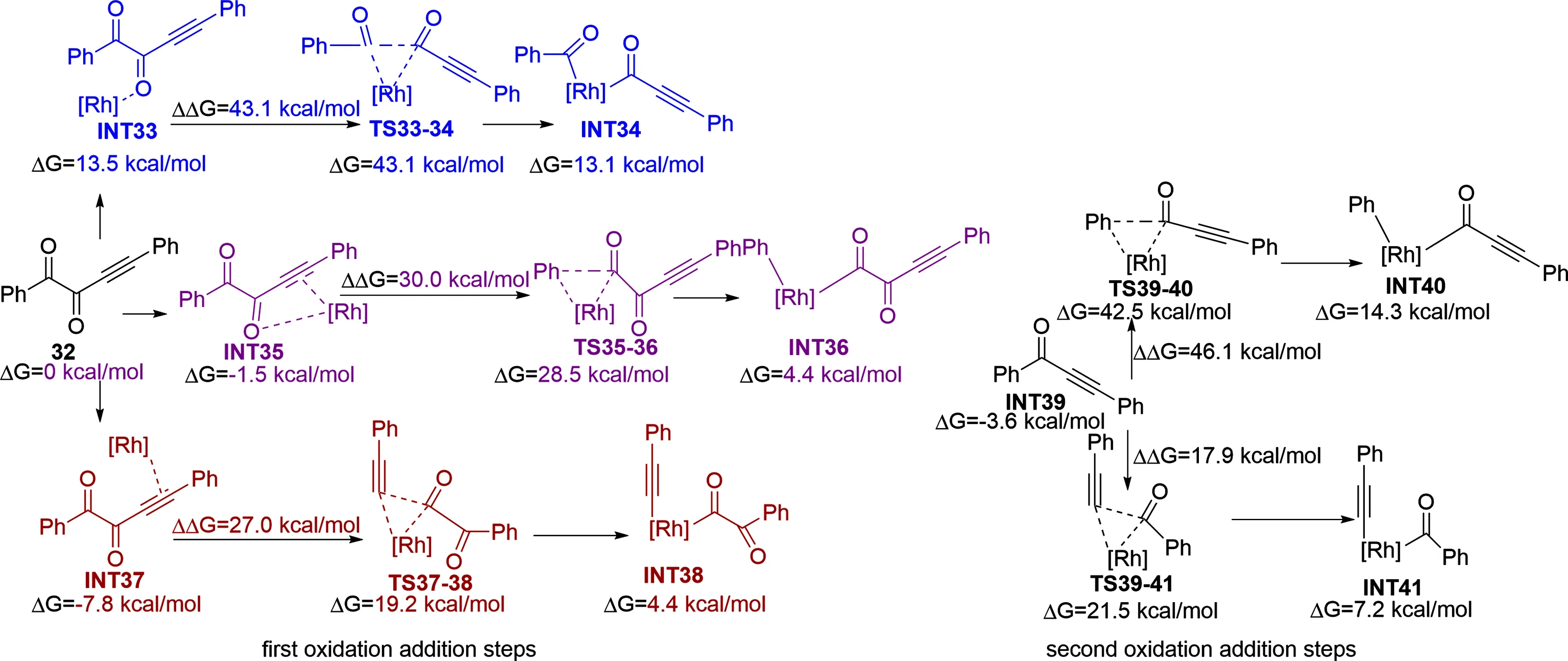
图7 铑催化二酮C—C键选择性活化步骤的势能剖面图(能量:kcal/mol)
2020年,Li等人[28]对铑催化烯丙基苯的烯基化反应中C—C键是通过β—C消除活化还是通过氧化加成活化,以及影响C—C键在2位或者5位活化的因素开展了DFT研究(如图8)。由计算结果可知,C—C键通过氧化加成或β—C消除发生活化需要克服的Gibbs 自由能垒分别为 44.9 kcal/mol(TS44)和 18.2 kcal/mol(TS45),表明在 90 ℃ 的实验条件下,通过氧化加成发生C—C键活化是不可能的,作者推测这可能与氧化加成途径产生的阳离子铑-氢配合物极其不稳定有关。此外,β—C消除途径中C—C键经2位活化(TS49-51)形成Rh-N配位中间体的过程仅需跨越16.0 kcal/mol Gibbs 自由能垒,经5位活化(TS48-50)时能垒为 27.1 kcalmol-1,考虑到前者的关键过渡态的相对能量均低于后者,则此反应倾向于得到2-位烯基化产物。说明吡啶基N的配位有利于稳定活化过渡态与中间体。计算结果与实验观察一致。
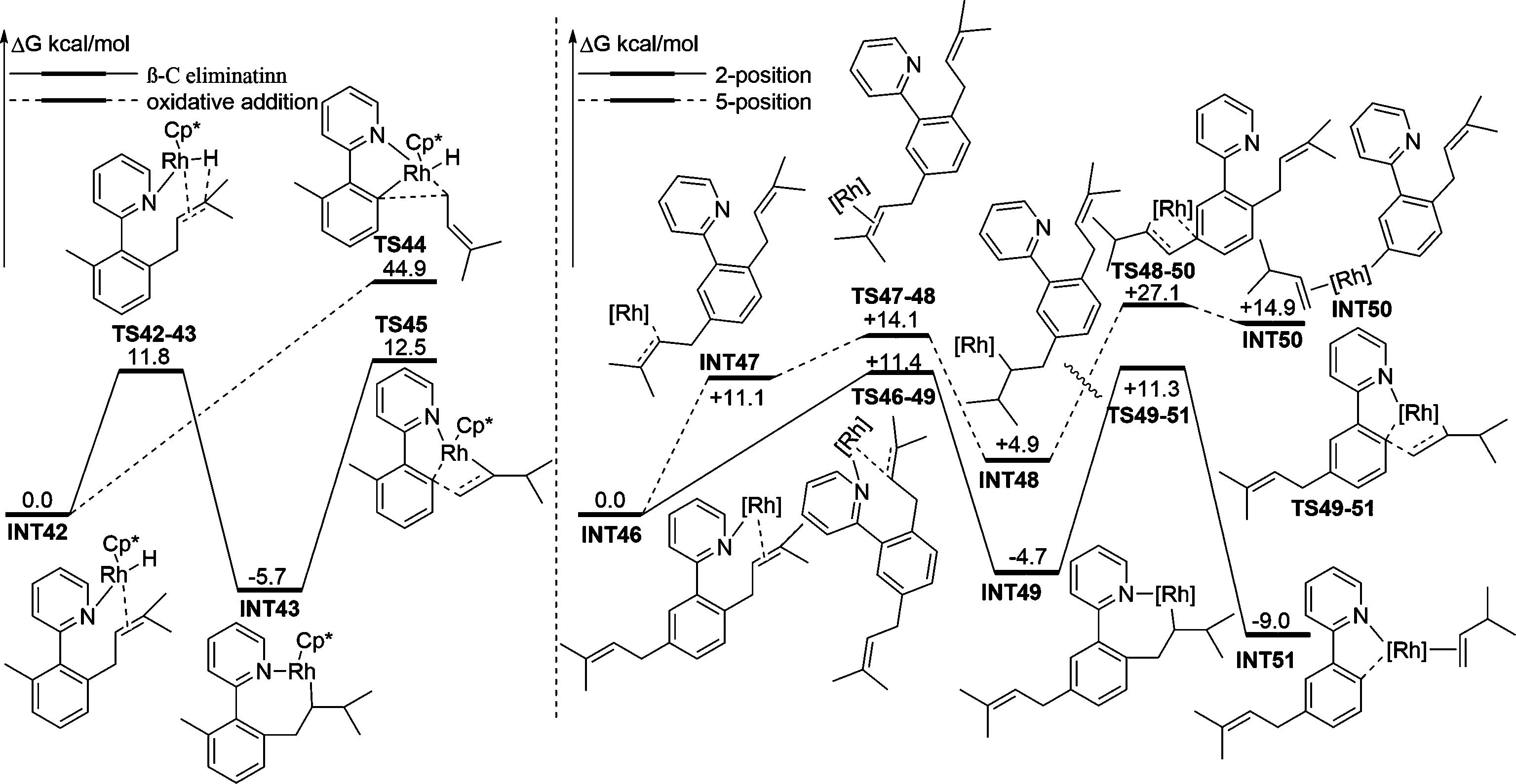
图8 铑催化烯丙基苯烯化反应中C—C键选择性活化的势能剖面图(能量:kcal/mol)
3 结论与展望
本文对近些年Rh催化C—C键选择性活化反应的密度泛函理论研究进展做了综述,对该类反应C—C键活化步骤的详细机理和区域选择性来源做出总结。多数C—C键活化反应的区域选择性主要与C—C键的稳定性和生成物质的稳定性有关,例如,空间位阻、弱配位、共轭、极性等。然而,现有的活化C—C键的方法仍不成熟,通常依赖于底物特性,如环应变、芳构化、螯合或协同辅助来诱导C—C键活化。相信经过更多化学工作者的努力,在计算化学的辅助下,这一领域的研究会变得更加完善。
