Radical cascade cyclization for the green and simple synthesis of silylated indolo[2,1-a]isoquinoline derivatives via visible light-mediated Si–H bonds activation
2023-02-18ZhenkiLeiFeiXueBinWngShijieWngYuXiYonghongZhngWeiweiJinChenjingLiu
Zhenki Lei ,Fei Xue ,Bin Wng ,Shijie Wng ,Yu Xi ,Yonghong Zhng ,Weiwei Jin,Chenjing Liu,b,∗
a State Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources,Key Laboratory of Oil and Gas Fine Chemicals,Ministry of Education& Xinjiang Uygur Autonomous Region,Urumqi Key Laboratory of Green Catalysis and Synthesis Technology,College of Chemistry,Xinjiang University,Urumqi 830017,China
b College of Future Technology,Institute of Materia Medica,Xinjiang University,Urumqi 830017,China
c Xinjiang Uygur Autonomous Region Product Quality Supervision and Inspection Institute,Urumqi 830011,China
Keywords:Indolo[2,1-a]isoquinolines Silyl radicals Green photocatalytic Simple photoinduced Silylated Hydrogen atom transfer EDA complex
ABSTRACT Photocatalytic and photoinduced silyl radicals cascade cyclization procedures for the green and simple preparation of fused tetracyclic skeleton silylated indolo[2,1-a]isoquinoline-6(5H)-ones from 2-aryl-N-acryloyl indoles with hydrosilanes are developed.The photocatalytic reaction is carried out with 9,10-dicyanoanthracene (DCA) as an organophotocatalyst and 3-acetoxyquinuclidine as hydrogen atom transfer(HAT) catalyst at room temperature under metal-and oxidant-free conditions.The keys to the success of photoredox-catalytic conversion include (1) the reductive quenching of DCA∗[E1/2(∗P/P–)=+1.97 V vs. SCE in MeCN] by 3-acetoxyquinuclidine (Ep=+1.22 V vs. SCE in MeCN),and (2) the thermodynamic feasibility of hydrogen atom abstraction from hydridic Si–H bond by electrophilic N+•.Particularly,the simple photoinduced cascade cyclization using (TMS)3SiH with 2-aryl-N-acryloyl indoles was exploited via an electron−donor−acceptor (EDA) complex under visible light irradiation.
Organosilicon molecules are evoked remarkable interests and explored deeply by synthetic chemists,pharmacologists and material scientists because of their conspicuous chemical,physical,and biological properties (I-V,Fig.1) [1–7].Especially,silicon as isostere of carbon in biomolecules have become new drug-like candidates in drug discovery [8].Classically,organosilicon derivatives were prepared by nucleophilic reactions of organometallic reagents with halosilanes [9–10],and transition-metal catalyzed cross-coupling of hydrocarbons or halogenated hydrocarbons with silylating reagents [11–20].Recently,there were two effective methods for the synthesis of organosilicon compounds from hydrosilanes [21] or Si–X (X=Si [22],B [23],COOH [24]) reagents with alkenes,alkynes and arenes [25].Among them,it was an atom-economical silylated approachviahomolytic cleavage of Si–H bonds in hydrosilanes to generate silyl radicals.The archetypical way was thermo-promoted peroxide decomposition to trigger silyl radicals [26].The second protocol was electron induced peroxide to initiate silyl radicals,the electron donors including transition-metal[27–29],TBAI [30] and photocatalyst [31].The third way was that alkali initiated silyl radicals [32].Although there have been significant advances,some methods suffered from harsh conditions or poor group compatibility,which would drive to find new strategies of triggering silyl radicals.

Fig.1. Special examples of silicon-containing active molecules (I-V) and indolo[2,1-a]isoquinoline derivatives (VI-VIII).
Photoredox catalysis [33–44] has appeared as a attractive protocol for silyl radicals generation via hydrogen atom transfer (HAT)of Si–H bonds [45–48].Fagnonietal.pioneered the tetrabutylammonium decatungstate (TBADT) as HAT photo-catalyst for trisubstituted silanes activation under phosphor-coated lamps irradiated by 310 nm [49].Unfortunately,due to the comparably high bond dissociation energies (BDEs) of Si–H andα–Si–C–H bonds in alkyl-substituted silanes (e.g.,triethylsilane) [50],the HAT process initiated simultaneously the cleavage of Si–H andα–Si–C–H bonds.So the selectivity of HAT catalyst for Si-H bonds was poor.For achieving the desired HAT of Si–H bonds,it is necessary that using “aggressive” radicals to break the BDEs of Si–H bonds [51],hence the process is thermodynamically favorable.For instance,an electrophotocatalytic HAT process was developed for silyl radicals generation using MeOH as HAT reagent [52].This work confirmed that hydrogen atom abstraction could be achieved by “aggressive” MeO•(BDEO–H=105 kcal/mol).Wuetal.developed an effective method for silyl radicals formation employing 3-acetoxyquinuclidine or triisopropyl-silanethiol as HAT reagent [53].This also showed the feasibility of hydrogen atom abstraction by“aggressive” N+•(BDEN+–H=100 kcal/mol) or S•(BDES–H=88.2 kcal/mol).What is more,because hydrogen is more electronegative than silicon in hydrosilanes,according to the polarity-matched effect [54],the electrophilic radical (e.g.,O•,N+•,S•) could be used to selectively abstract hydrogen of Si–H bonds in hydrosilanes rather thanα–Si–C–H bonds.
(TMS)3SiH was an ideal reagent in radical chemistry,which was used in many tris(trimethylsilyl)silylation or conversion processes.Because (TMS)3SiH has noα–Si–C–H bond and BDESi–His relatively low,(TMS)3Si•radical could be initiatedviahydrogen atom abstraction by HAT reagent,single electron oxidation of (TMS)3SiH by PC∗and then deprotonation [55],phosphor coating fluorescent lamp and UV light irradiation [49,56-58],etc.[59,60].However,only two examples were reported that (TMS)3Si•radical was producedviaformation an electron−donor−acceptor (EDA) complex with alkyl or aryl halide to abstract halogen under visible light irradiation [61,62].
Indolo[2,1-a]isoquinolines containing the tetracyclic skeleton are widely found in bioactive and pharmaceutical molecules(VI-VIII,Fig.1) [63–69].Due to the potential of silicon incorporation in drug discovery,it is of great significance for the synthesis of silylated indolo[2,1-a]isoquinoline compounds.So far,few cases of synthesis have been reported,including Cu(acac)2/TBPB-initiated triethylsilyl radical cascade cyclization(Scheme 1a) [70],cerium-electrophotocatalyzed methoxyl radicalmediated triethylsilyl radical cascade cyclization (Scheme 1b) [52],and palladium-catalyzed cascade cyclization with hexamethyldisilane [71] or Me3SiSiMe2(OnBu) (Scheme 1c) [72].Despite significant advances,the fly in the ointment was that these examples were heating conditions,besides,there were one or more shortcomings,such as stoichiometric oxidant,poor atom economy,preactivation of substrates and expensive transition-metal catalysts.
Taking into account the above aspects and our continuing interest in the preparation of heterocyclic molecules under visible light conditions [73–79],herein we report photocatalytic HAT selectively initiated silyl radicals cascade cyclization for the synthesis of silylated indolo[2,1-a]isoquinoline compounds.In addition,the simpler and greener cascade cyclization using (TMS)3SiH was exploitedvianovel EDA complex,the tris(trimethylsilyl)silylated indolo[2,1-a]isoquinolines can be obtained successfully under visible light irradiation even in the absence of photocatalyst and HAT catalyst (Scheme 1d).
Preliminary research was investigated by 1-(2,3-diphenyl-1Hindol-1-yl)-2-methylprop-2-en-1-one (1a) and triethyl-silane (2a)as model reaction substrates,and the outcomes were summarized in Table 1 and Tables S1-S5 (Supporting information).After screening these detailed conditions,it was found that the optimal choice including 0.1 mmol of1a,10 equiv.of2a,10 mol% of PC1,and 12.5 mol% of HAT cat.1in 2 mL dry MeCN under 10 W blue LEDs irradiation at room temperature for 30 h.And the target product3awas isolated with a yield of 70% under the optimal conditions.
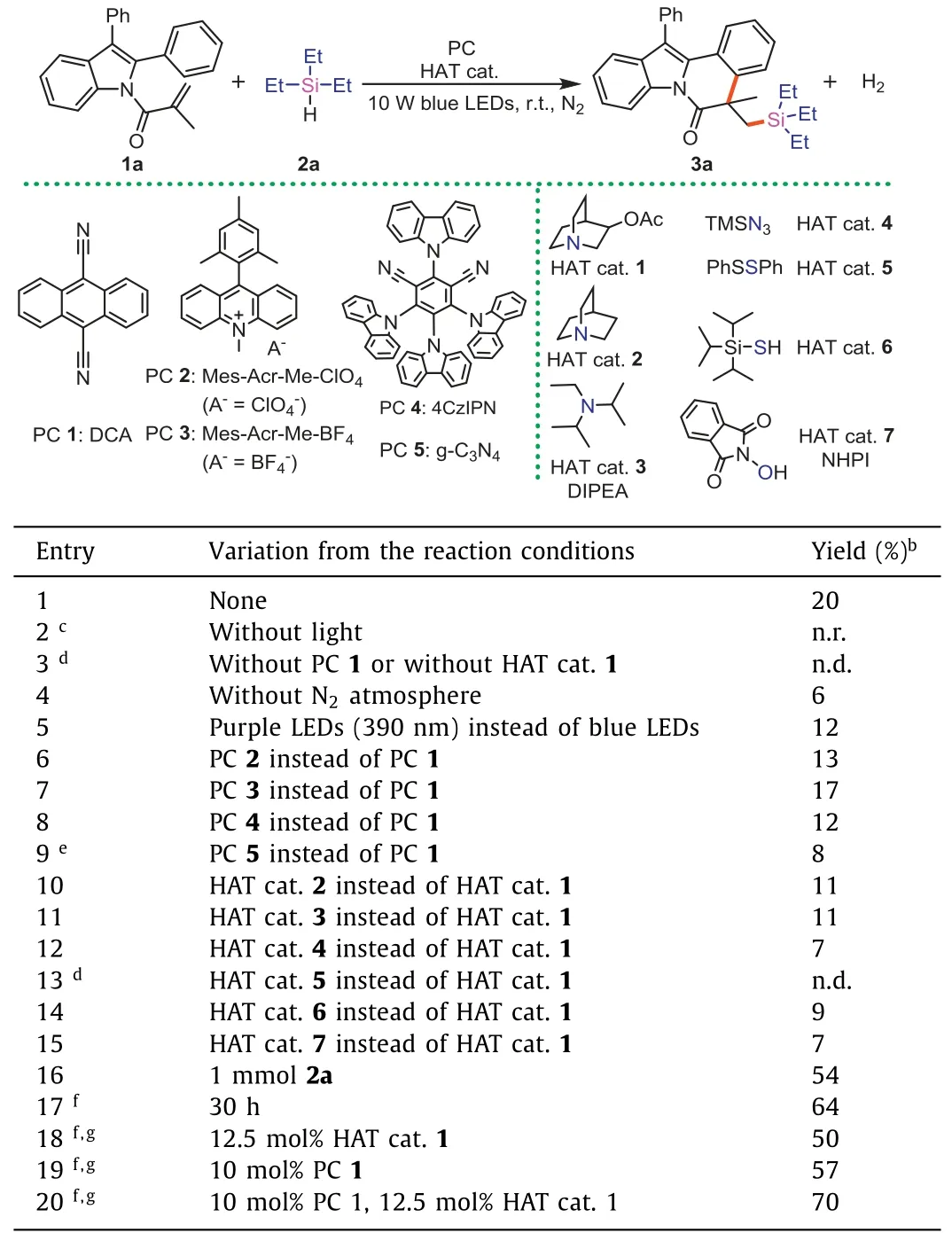
Table 1 Optimization of reaction conditions.a
Having confirmed the optimal reaction conditions,we next evaluated the scope of 2-aryl-N-acryloyl indoles (Scheme 2).For example,substrates containing electron-withdrawing groups (F-,Cl-,CN-,and CF3O-) at C5-position of indole ring could yield the desired products3b-3ein 42%-53% yields.Electron-donating groups (Me-,Et-,andiPr-) were also good compatibility,giving the expected products3f-3hwith yields of 53%-65%.

Scheme 2. Scope of 2-aryl-N-acryloyl indoles.Reaction conditions: 1 (0.1 mmol), 2a (1 mmol),PC 1 (10 mol%),HAT cat. 1 (12.5 mol%),dry MeCN (2 mL),10 W blue LEDs,N2,r.t.,30 h.Isolated yields.
We next inspected the scope of hydrosilanes (Scheme 3).Under optimal conditions,arylsubstituted silanes such as triphenylsilane,diphenylmethylsilane and phenyldimethylsilane could be gave the desired products3k(proved by X-ray crystallography),3land3min 42%-55% yields.Trialkylsilanes showed good selectivity to afford the desired products3n-3qin 54%-66% yields,whereas the competing reaction of the C–H adjacent to silicon was not observed.Moreover,2-aryl-N-acryloyl indoles and hydrosilanes could also combine freely to make new products,such as substrates1cand triphenylsilane worked smoothly.But the synthesis of triethoxysilylated product3swas failed,presumably because the BDE of Si-H bond in triethoxysilane is high.
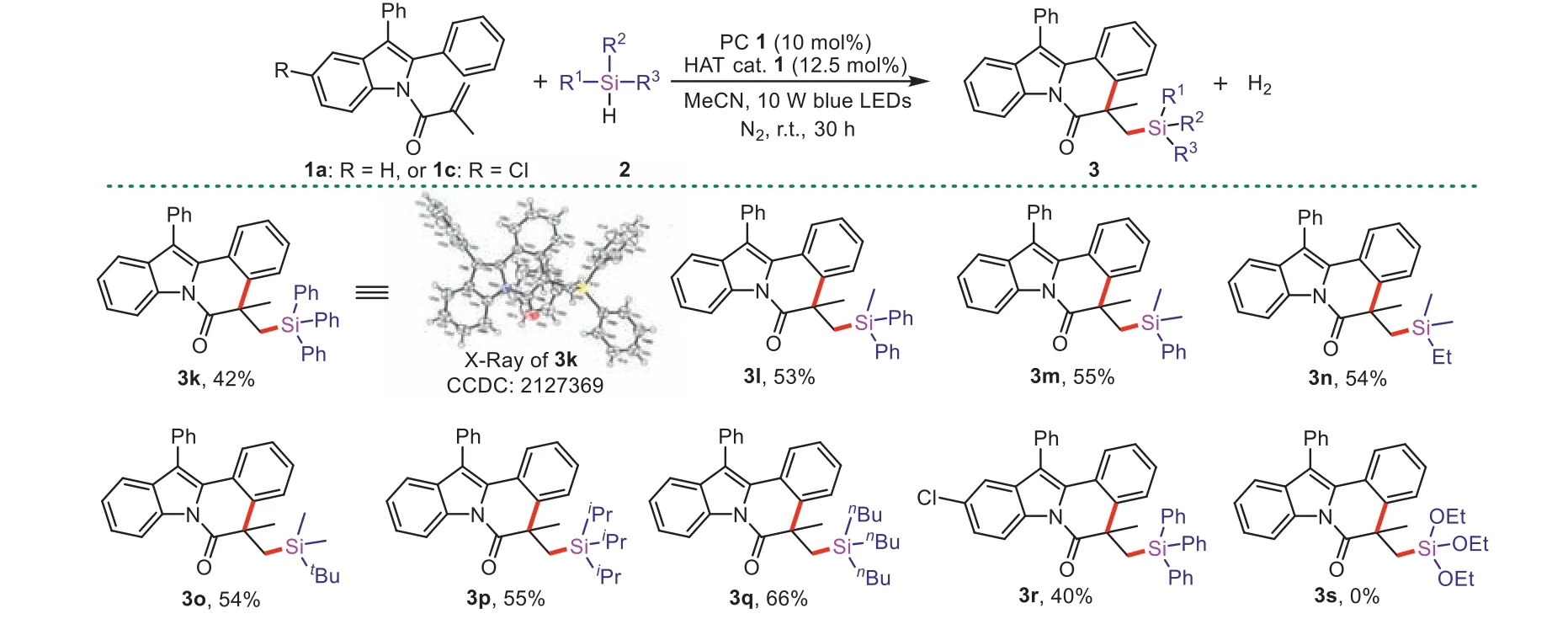
Scheme 3. Scope of hydrosilanes.Reaction conditions: 1a or 1c (0.1 mmol), 2 (1 mmol),PC (DCA,10 mol%),HAT cat. 1 (12.5 mol%),dry MeCN (2 mL),10 W blue LEDs,N2,r.t.,30 h.Isolated yields.
We further found that 2-aryl-N-acryloyl indoles could successfully react with (TMS)3SiH under 10 W blue LEDs irradiation(Scheme 4).The reaction conditions were optimized and displayed in Tables S6-S9 (Supporting information).Under the optimized reaction conditions,expected products3t(confirmed by X-ray crystallography),3u-3w,3yfrom F-,Cl-,Br-,and CF3O-groups located at C4-or C5-position of indole ring could be gained with moderate yields of 50%-66%.The 6,7-dichloro substituted product was also obtained,although the product3xwith a low yield.Electrondonating groups (5-methyl,5-isopropyl,4,6-dimethyl) were tolerated,giving the desired products (3aa,3ab,3ac) in 58%-69% yields.
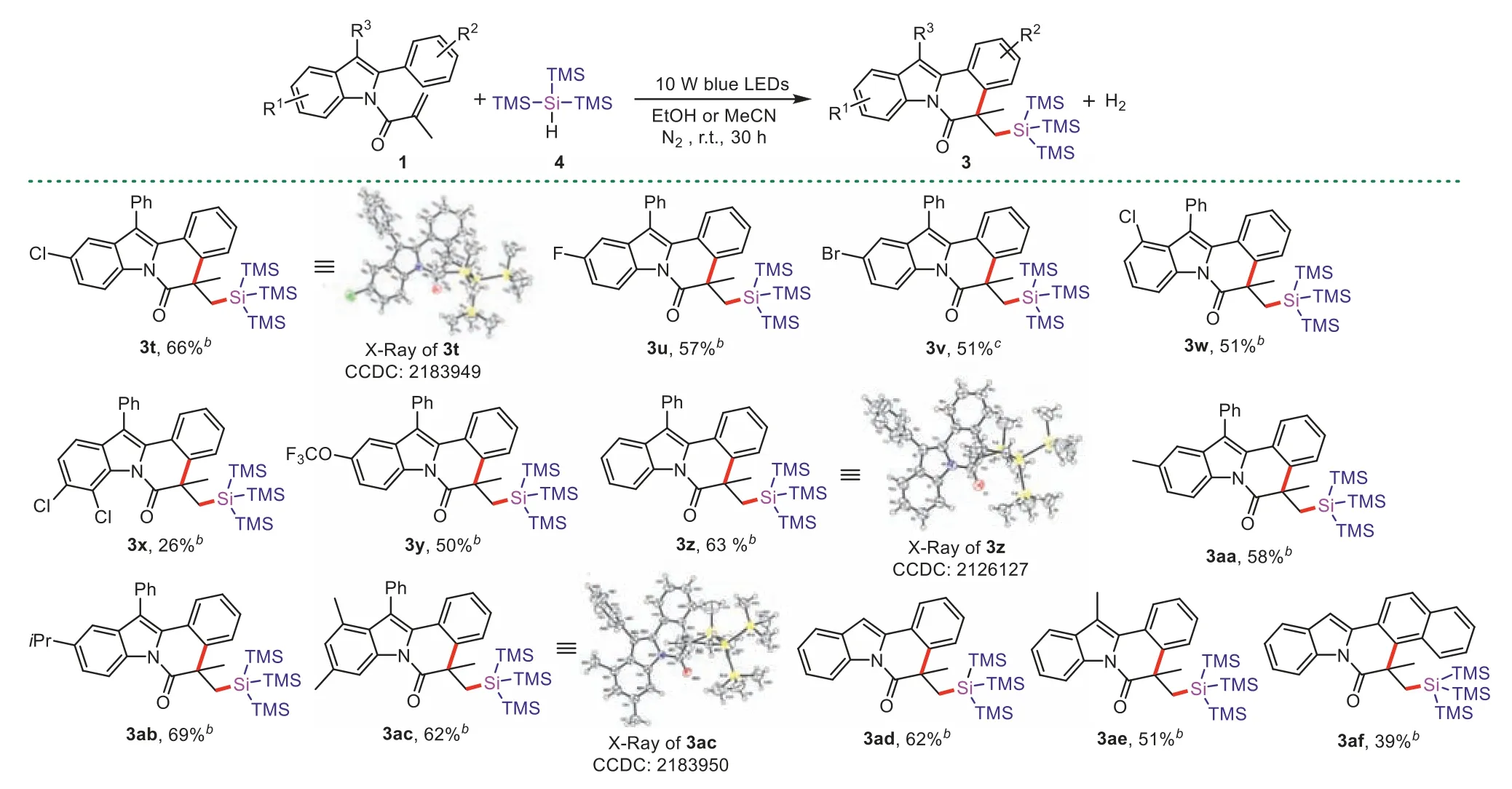
Scheme 4. Scope of reaction between 2-aryl-N-acryloyl indoles and (TMS)3SiH.Reaction conditions: 1 (0.1 mmol), 2 (1 mmol),dry EtOHb or MeCNc (1 mL),10 W blue LEDs,N2,r.t.,30 h.Isolated yields.
The synthetic application of compound3twas presentedviafurther transformations (Scheme 5).Reduction of3twas investigated,the carbonyl group could be reduced to obtain compound5in 60% yield.Desilylation of the tri(trimethylsilyl)silyl group in3twas performed with Bu4NF (TBAF) to give a disilane product6in 38% yield under microwave irradiation.
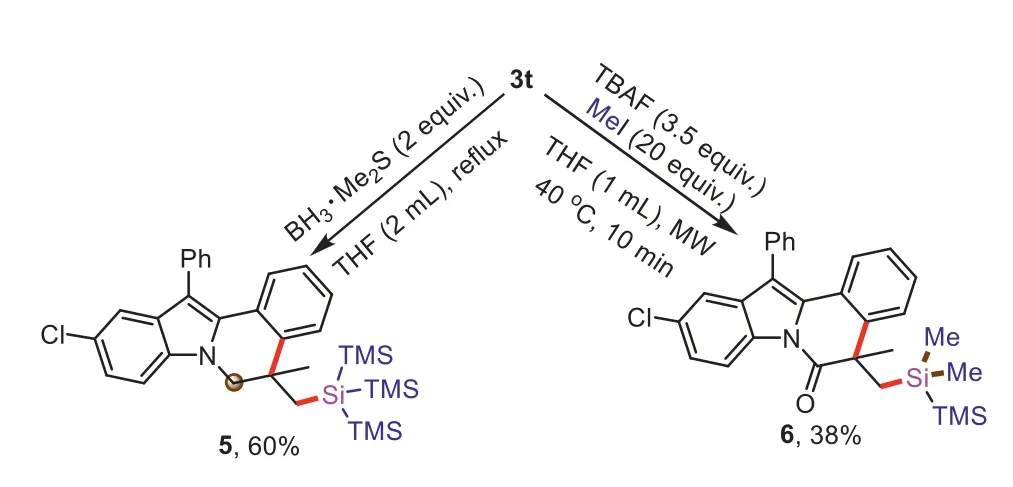
Scheme 5. Synthetic transformations of 3t.
To explore the reaction mechanism,control experiments were implemented.The photocatalytic reaction was completely restrained while adding 2 equiv.of TEMPO under the standard conditions.Similarly,the reaction was conducted in the presence of BHT or 1,1-diphenylethane,the yield of3awas significantly reduced(Fig.2a),which demonstrated that a radical process might be involved.Furthermore,the BHT-trapped product7was detected by HRMS.Besides,no reaction happened while employing deuterated diphenylmethyl silane (Ph2MeSiD) (Fig.2b).Such a significant kinetic isotope effect (KIE) suggested that the rate-determining step involved the cleavage of the Si–H bonds.The H2was monitored by H2detector and GC under standard conditions (Fig.S4 in Supporting information).“On/off” experiments indicated that visible light played an important role (Fig.2c).In addition,Stern-Volmer fluorescence quenching experiments demonstrated that the excitedstate DCA∗was quenched by HAT cat.1(Figs.2d and e) through single electron transfer (SET) process.
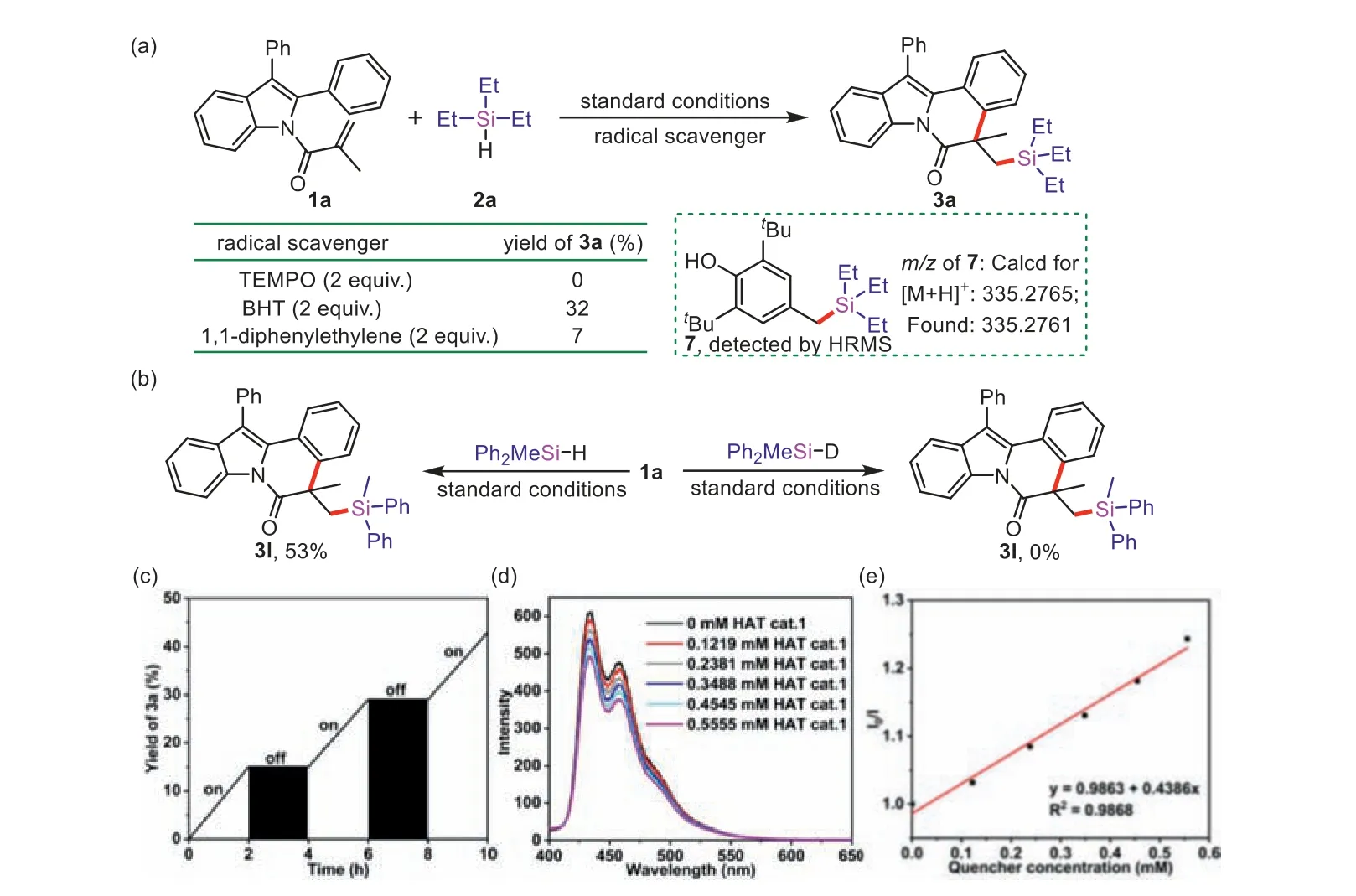
Fig.2. Mechanistic investigations.(a) Radical trapping experiment;(b) Investigation of the KIE;(c) ON/off experiments;(d) Fluorescence quenching experiment (DCA (1 μmol/L in MeCN) with different concentration of HAT cat. 1 were irradiated by 365 nm);(e) Stern-Volmer plot of HAT cat. 1.
On the basis of the above mechanistic investigations,a plausible mechanism was proposed,as depicted in Fig.3a.Initially,DCA was excited to produce the long-life photoexcited-state DCA∗(t=14.9 ns) [80].The reductive quenching of DCA∗[E1/2(∗P/P–)=+1.97 Vvs.SCE in MeCN] [81] by 3-acetoxyquinuclidine (Ep=+1.22 Vvs.SCE in MeCN) [82] leaded to the radical anionAand a radical cation intermediateB.Due to its high electrophilicity,quinuclidinium radical cationBselectively abstracted the hydrogen atom from the more hydridic Si-H bonds of hydrosilanes to produce the corresponding silyl radicalC,as well as quinuclidinium cationD.This abstraction event should be thermosdynamically favorable because the BDESi–Hof hydrosilanes was up to 94.6 kcal/mol and the BDEN+–Hin quinuclidinium cationDwas 100 kcal/mol.Subsequently,a carbon-centered radical intermediateEwas generated by the addition of the silylic radicalCto the C=C bond of the indole substrate1a,then the radical intermediateEwas cyclizedvia6-exo-trigpathway to afford the radical intermediateF.Afterwards,the single-electron oxidation process ofFandA[E1/2(P/P–)=–0.97 Vvs.SCE in MeCN] [81] occurred simultaneously to give the cationGand regenerate the DCA.In this process,there was sufficient driving forces to yield H2through the reduction of two protons[83,84].Finally,the deprotonation of the cationGgave the silylated product3.

Fig.3. Proposed reaction mechanism for the formation of silylated indolo[2,1-a]isoquinoline-6(5H)-ones.
Particularly,controlled experiments were performed to study the mechanism of the reaction that1creact with (TMS)3SiH (4)under the photoinduced conditions in Scheme 4.The product3twas fully inhibited when 2 equiv.of TEMPO was added.Meanwhile,the BHT or 1,1-diphenylethane was added,the yield of3twas decreased (Section 4.1 in Supporting information),which demonstrated a radical pathway might also be involved.With the reaction proceeded,the color of the solution became yellow gradually,the H2was observed clearly and the concentration increased gradually (Fig.S5 in Supporting information).Subsequently,“on/off” LED irradiation experiments also showed that visible light played a key role in the reaction (Fig.S6b in Supporting information).We tested optical absorption of the EtOH solution of1cand4,it was not observed the red-shift or new absorption peak in the UV-vis absorption spectra (Fig.S8 in Supporting information).Next,we conducted1H NMR experiments,and the chemical shift of4shifted downfield with increasing amounts of1c(Fig.4).These experimental results showed the formation of EDA complexes from 2-aryl-N-acryloyl indoles1with4.So the (TMS)3Si•radical could be formed by excited EDA complexes and an energy transfer under blue LEDs irradiation (Fig.3b).The remaining mechanism including (TMS)3Si•radical addition and cascade cyclization was the same as described in Fig.3a.It was worth noting that the single electron oxidation ofF’was accompanied by the reduction of protons.

Fig.4. 1H NMR experiments between 1c and 4.
In summary,we developed green and simple photocatalytic and photoinduced silyl radicals cascade cyclization protocols for the synthesis of silylated indolo[2,1-a]-isoquinoline-6(5H)-ones.The photocatalytic procedure was conducted in the presence of DCA as photocatalyst and 3-acetoxyquinuclidine as HAT catalyst.To implement the desired silylation,the reductive quenching of DCA∗by HAT catalyst,the abstraction hydrogen using “aggressive” radical were necessary,and the polaritymatched effect was also the key factor in success.The simple photoinduced method achieved the straightforward preparation of tris(trimethylsilyl)silylated indolo[2,1-a]-isoquinoline-6(5H)-onesviaEDA complex.These two facile and greener procedures have the advantages including high atomic economy,H2as by-product,metal-free,oxidant-free,easy operation,and mild reaction conditions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was supported by the Tianshan Talents Program for Leading Talents in Science and Technology Innovation (No.2022TSYCLJ0016),the National Natural Science Foundation of China(Nos.21961037 and 22201241),the Program for Tianshan Innovative Research Team of Xinjiang Uygur Autonomous Region (No.2021D14011),the Graduate Innovation Project of Xinjiang Uygur Autonomous Region (No.XJ2021G036),the Key Program of Natural Science Foundation of Xinjiang Uygur Autonomous Region(No.2022D01D06),and the Natural Science Foundation of Xinjiang Uygur Autonomous Region (Nos.2021D01E10 and 2022E01042).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108633.
杂志排行

Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers