Direct synthesis of unnatural amino acids and modifications of peptides via LADA strategy
2023-02-18YunqiLiuJunliangZhouZhankuiSun
Yunqi Liu ,Junliang Zhou ,Zhankui Sun
a Shanghai Frontiers Science Center for Drug Target Identification and Drug Delivery,School of Pharmaceutical Sciences,Shanghai Jiao Tong University,Shanghai 200240,China
b Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs,Pharm-X Center,School of Pharmaceutical Sciences,Shanghai Jiao Tong University,Shanghai 200240,China
c AI Pharma Center,Zhangjiang Institute for Advanced Study,Shanghai Jiao Tong University,Shanghai 201203,China
d Shanghai Artificial Intelligence Laboratory,Shanghai 200232,China
Keywords:Unnatural amino acids Peptide modifications Desulfurization Cysteine Radical addition
ABSTRACT Unnatural amino acids (UAAs) have broad applications in pharmaceutical sciences and biological studies.Current synthetic methods for UAAs mainly rely on asymmetric catalysis and often require several steps.There is a lack of direct and simple methods.To address this challenge,we designed the LADA (labelingactivation-desulfurization-addition) strategy: selective labeling and activation of cysteine residues,the photocatalytic desulfurization and the subsequent radical addition to alkenes.Although composed of two steps,it is one-pot synthesis and has advantages such as high functional group tolerance,biocompatible reaction condition,and retained stereochemistry.This highly efficient strategy was successfully applied in the direct synthesis of unnatural amino acids and modifications of peptides with more than 50 examples.
Unnatural amino acids (UAAs) are valuable building blocks in organic synthesis,medicinal chemistry and materials science [1].UAAs also find wide application in drug discovery,peptide and protein labeling,and protein engineering [2].Due to its high importance,many synthetic methods have been developed [3–12].Most known methods rely on asymmetric phase-transfer catalysis,transition-metal catalysis and Strecker reaction,which often require multi-step synthesis.Despite huge advance,methods which could be used for the direct chemical modification of peptides and even proteins are still in great needs.
Natural amino acids provide perfect handles for further elaboration,such as thiol group in cysteine [13–16].B.G.Davis group converted cysteine residue to dehydroalanine (Dha) and utilized Dha for further modification of proteinsviaradical addition reactions [17,18].However,the stereochemistry was hard to control(Scheme 1).N.J.Mitchell and co-workers developed visible-lightmediated desulfurative C–C bond forming reaction which enabled the site selective installation of modified sidechains into peptides and proteins [19].But the efficiency of this strategy needed to be improved.D.Niuetal.also discovered a radical approach to convert C-S bonds of cysteine into C–C bonds for the synthesis of UAAs[20].The requirement for the pre-installation of activation group may limit its application.

Scheme 1. Current methods for synthesis of UAAs based on cysteine residue and the proposed LADA strategy.
Based on our previous work [21–24],we proposed a novel strategy which we called LADA (labeling-activation-desulfurizationaddition) strategy (Scheme 1).This two-step strategy includes the selective labeling and activation of cysteine residues,the photocatalytic desulfurization to generate radical intermediates and the subsequent radical addition to alkenes to deliver UAAs and modified peptides as the products.Although composed of two steps,it is actually one-pot synthesis and could be handled easily.The advantages of this LADA strategy include excellent functional group tolerance,high efficiency,broad scope,etc.Meanwhile,the configuration of the modified amino acids remains untouched.All of these features bestow this LADA strategy great potential to be further applied in chemical modifications of proteins.Herein we report the realization of this LADA strategy for the synthesis of UAAs and modifications of peptides.
We began our investigation by using cysteine derivativeAas model substrate and examined different conditions.We found that under basic condition dihydro-1H-imidazole saltB1could effi-ciently activate thiol group in cysteine within one hour and afforded intermediateC1,which reacted with silyl enol etherD1upon photocatalysis.Thus,under standard condition,product1was isolated in 89% yield (>99%ee,Table 1,entry 1).To our surprise,with imidazole saltB2andB3,the yields were much lower,indicating the importance of counter ions (Table 1,entries 2 and 3).We also evaluated solvents other than MeCN/H2O,such as DMSO/H2O,THF/H2O,however,the yields were lower (Table 1,entries 4–6).At last,we tested different photocatalysts and Ir(ppy)3proved to be the best (for a detailed account of the optimization studies,see Table S1 in Supporting information).Further control experiments revealed that no reaction happened without photocatalyst or blue LEDs (Table 1,entries 7 and 8).
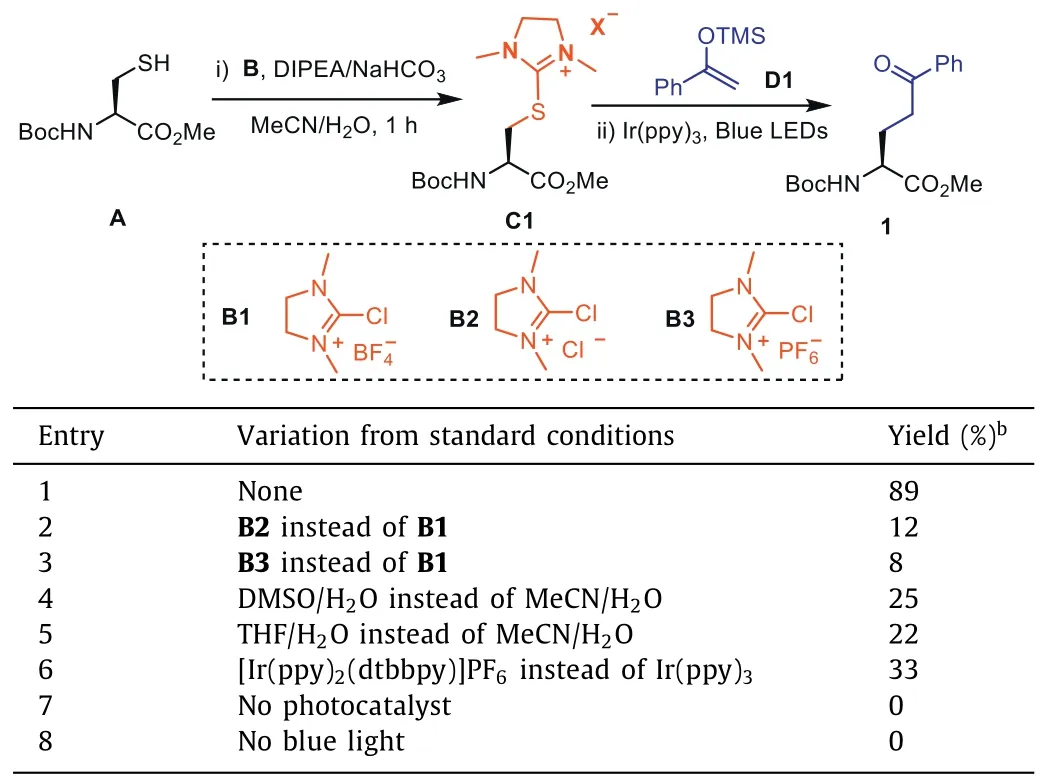
Table 1 Optimization of the reaction conditions.a
Having established the optimized conditions,we started to probe the scope of this transformation.We first evaluated this method with different vinyl ethers.Silyl enol ethers such as TMS and TBS afforded the products in good to excellent yields (Table 2,entries 1 and 2).Vinyl acetate and vinyl carbonate also delivered the products in 86% and 65% yields (Table 2,entries 3 and 4).
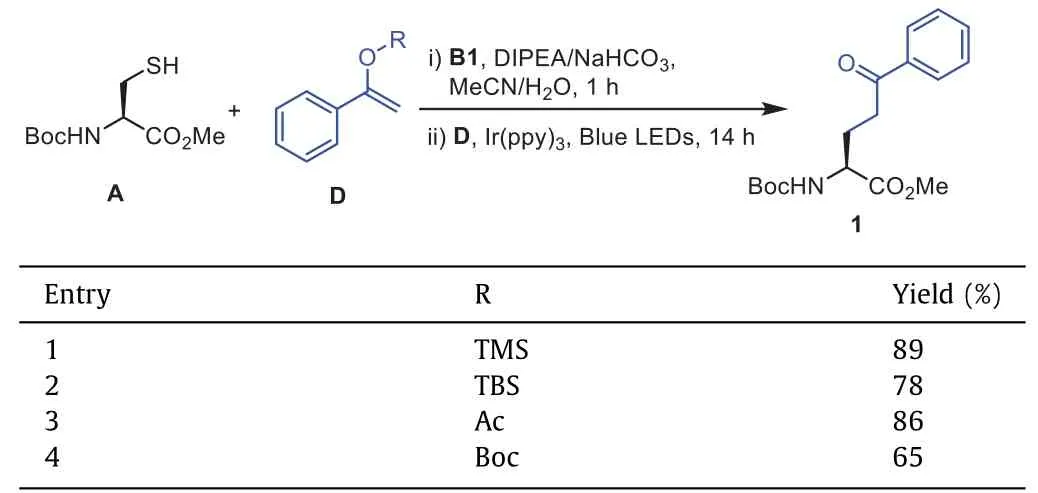
Table 2 Substrate scope of vinyl ethers.a
We then started to evaluate the scope of silyl enol ethers as listed in Scheme 2.For phenyl bearing electron donating groupssuch as phenyl,tert–butyl,andn-octyl,the products were isolated in excellent yields (2–4).One notable example was the azide derivative,which furnished the product in 73% yield (5).For strong electron donating groups,such as alkyloxy groups,the yields were generally high (6–12).Interesting examples include the one bearing alkyne and cyclobutane ester (11–12).Electron withdrawing groups were tolerated as well and delivered the products in good yields.For examples,phenyl with halogen atoms such as F,Cl,Br,and I,afforded the products in 59%−87% yields (13–16).Strong electron withdrawing groups including CF3,ester and pyridine also provided the products in good yields (17–19).For cyclohexyl and cycloheptyl silyl enol ether,the products were isolated in 65% and 69% yields with 1:1 diastereoselectivity (20,21).However,alkyl silyl enol ethers did not provide the products.

Scheme 2. Substrate scope of silyl enol ethers.Reaction conditions: i) A (0.2 mmol), B1 (0.22 mmol),DIPEA (0.2 mmol),NaHCO3 (0.44 mmol),MeCN/H2O (4 mL/0.05 mL),room temperature for 1 h.ii) Ir(ppy)3 (1.5 mol%), D (0.5 mmol),blue LEDs,room temperature,14 h,N2.Isolated yield.
We further expanded this reaction to alkenes bearing terminal carboxyl group or hydroxyl group (Scheme 3).For example,4-phenylpent-4-enoic acid was efficiently converted into 1,4-butyrolactone derivative22in 74% yield.Other alkene-acids were also compatible and successfully transformed into the corresponding products in good yields (23–28).Both electron donating groups and electron withdrawing groups were well tolerated.Interestingly,when phenyl ring was used to bridge alkene and acid,the yields were generally higher (29–32).For instance,compared with 74% yield for product22,product30was isolated in 88% yield.While for product31,the yield was increased to 80% as compared to 73% for derivative28.When alkenes with terminal hydroxyl groups were subjected to the standard conditions,the reactions also worked smoothly and the products were isolated in 61%−71%yield (33–36).

Scheme 3. Substrate scope of alkenes.Reaction conditions: (i) Bz-Cys (0.2 mmol), B1 (0.22 mmol),DIPEA (0.2 mmol),NaHCO3 (0.44 mmol),MeCN/H2O (4 mL/0.05 mL),room temperature for 1 h.(ii) Ir(ppy)3 (1.5 mol%), E (0.26 mmol),NaHCO3 (0.20 mmol),blue LEDs,room temperature,40 h,N2.Isolated yield.
In an effort to establish the generality of this protocol,we did late-stage modifications of natural products and commercial drugs.Thus,under standard condition,Clofibrate,Celestolide,Tonalide,Ibuprofen and Ciprofibrate derivatives were successfully transformed into unnatural amino acids in good to excellent yields(Scheme 4,37–41).

Scheme 4. Late-stage modifications of commercial drugs.Reaction conditions: (i) A (0.2 mmol), B1 (0.22 mmol),DIPEA (0.2 mmol),NaHCO3 (0.44 mmol),MeCN/H2O(4 mL/0.05 mL),room temperature for 1 h.(ii) Ir(ppy)3 (1.5 mol%), D (0.5 mmol),blue LEDs,room temperature,14 h,N2.Isolated yield.
To further demonstrate the potential of this strategy,we performed direct chemical modifications of cysteine derivatives(Scheme 5).Under standard condition,tert–butyl ester and Fmocprotected cysteine provided the products in 72% and 65% yields(42,43).We were pleased to find out this reaction worked smoothly with peptides and the products were delivered in good to excellent yields.For example,Ala-Cys,Val-Cys,Pro-Cys,and Phe-Cys-were converted to the products in 60%−88% yields (44–47).For Glu-Cys,the modified product was isolated in 63% yield (48).As expected,Tyr-Cys-and Ser-Cys-with free hydroxyl groups proceeded efficiently and delivered the products in 73% and 70% yields(49,50).Trp-Cys-also afforded the product in good yield (51).Fro peptides which have basic amine groups,such as Lys-Cys-and Arg-Cys,the reactions also worked smoothly (52,53).However,for His-Cys,the yield was unsatisfactory (54).We also tested on tripeptides,such as Gly-Pro-Cys-and Val-Pro-Cys.The products were isolated in 82% and 69% yields (55,56).However,for unprotected peptides,this reaction didn’t work well.

Scheme 5. Direct chemical modifications of cysteine derivatives.Reaction conditions: (i) AA (0.2 mmol), B1 (0.22 mmol),DIPEA (0.2 mmol),NaHCO3 (0.44 mmol),MeCN/H2O(4 mL/0.05 mL),room temperature for 1 h.(ii) Ir(ppy)3 (1.5 mol%),D (0.5 mmol),blue LEDs,room temperature,14 h,N2.Isolated yield.
To better understand the mechanism,we did control experiments as illustrated in Scheme 6A.When 2,2,6,6-tetramethyl-1-piperidyloxy (TEMPO) was added to the reaction under standard condition,we did not observe the formation of product60.Instead,we isolated dehydroalanine57and imidazolidine-2-thione58.At the same time,we detected the formation of59by HRMS.We then did radical clock experiments.We were able to obtain product61in 39% yield.We also performed Stern-Volmer experiments and found out intermediateC2could quench the photocatalyst under nitrogen atmosphere (Fig.S3 in Supporting information).

Scheme 6. Mechanistic study and proposed mechanism.
Based on the above experiments and our understanding,we proposed a plausible mechanism as shown in Scheme 6B.Upon photo-irridation,intermediateCis reduced to generate radical intermediateI.ViaC-S bond cleavage,radicalIbreaks into imidazolidine-2-thione58and radical speciesII.RadicalIIadds to silyl enol etherDand produces radical intermediateIII,which is further oxidized by photocatalyst IrIVto afford cationIV.At the same time,photocatalyst IrIIIis regenerated.In the final step,cationIVaffords1as the product upon loss of a TMS+.
In conclusion,we have successfully developed the LADA strategy.This strategy utilizes imidazole salt to selectively label and activate cysteine residues toin-situgenerate photo-reactive intermediate.Upon photocatalysis,this activated intermediate will generate carbon centered radicalviaC-S bond cleavage.This radical could add to different alkenes to deliver UAAs and modified peptides.This strategy has obvious advantages,such as excellent functional group tolerance,high efficiency,easy to operate,etc.Meanwhile,the configuration of the modified amino acids remains untouched.This LADA strategy provides simple yet robust solutions for the synthesis of UAAs and the chemical modification of short peptides,and will certainly have broad applications in organic synthesis,medicinal chemistry and other fields.
Note
A patent application based on the subject matter of this manuscript has been filed to China National Intellectual Property Administration (Nos.CN202210780183.8 and CN202310280695.2).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We thank Shanghai Jiao Tong University for financial support.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108553.
杂志排行

Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers