原子尺度钴基氮碳催化剂对析氧反应的构效关系的研究
2023-02-17吴明亮章烨晖付战照吕之阳李强王金兰
吴明亮,章烨晖,付战照,吕之阳,李强,*,王金兰,*
1东南大学物理学院,南京 211189
2东南大学机械工程学院,江苏省微纳生物医疗器械设计与制造重点实验室,南京 211189
1 Introduction
With the increase in fossil oil consumption and worldwide attention to the environment, developing sustainable, clean, and renewable energy has become one of the most critical challenges1–3. Storing excess electrical energy in chemical bonds, such as hydrogen production by water splitting driven by electricity, is a promising strategy to alleviate the energy crisis4.In the water splitting process, oxygen evolution reaction (OER)as a half-reaction, involves a multi-step electron transfer process,thermodynamic climbing, and slow kinetic process, which dramatically limits the total water splitting efficiency5,6. At present, nanomaterials based on IrO2/RuO2are generally considered to be the most advanced OER electrocatalysts7. Still and all, their large-scale application is hindered by high cost, low natural reserves, and relatively poor durability8. Thus,exploiting OER electrocatalysts with high-efficiency and low cost is significantly indispensable9–11.
Recently, transition metal-based (TM, Co, Ni, Fe) M-N-C catalysts have attracted extensive studies due to their low cost,high stability/conductivity, and flexible electronic structure of the active center12–14. Compared with traditional metal oxides bulk, nanoparticles, M-N-C catalyst with the concept of single atom catalysts (SACs) provides the most outstanding atomic efficiency15,16. Moreover, with the help of advanced characterization techniques like X-ray absorption near edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy techniques17, the fine structure of M-N-C catalysts can be further explored. The characterization of this kind of structure can bridge the experiment and theory,which is beneficial to further exploring the active origin of the reactive sites. However, the activity of electrocatalytic reactions is highly structurally sensitive, and slight changes in the coordination environment can lead to large changes in the activity18,19. Meanwhile, the dynamic evolution of the structure during the reaction can also lead to changes in reactivity20. This makes it difficult to understand the role played by the active sites to modulate the activity, which further limits the improvement and new design of active catalysts. Among the widely studied metal SACs, the cobalt-based M-N-C catalyst show high oxygen evolution reaction activity. For example, Zhang et al. fabricated an atomically dispersed cobalt- and nitrogen-codoped graphene catalyst with the configurations of Co-N4and Co-N2C2, showing an ultra-low onset overpotential of approximately 210 mV21.Zeng et al. theoretically predicted that the SACs with a Copyrrole-N4center could efficiently catalyze OER compared with pyridine nitrogen coordination22. Lu et al. found that cobalt nanoparticles@N-doped carbon composite exhibited a low onset overpotential of 300 mV for OER, which is similar to the performance of reported single-atom Co-N-C catalysts23.Therefore, the Co-N-C structure is chosen as an example system in this work to study the structure-activity relationship12,24–26.
In this study, we investigate the Co-N-C catalysts with different configurations (single atom, diatoms, and cluster) to determine the source of OER activity by free energy calculations and polarization curve simulations. The results show that the coordination environment of metal atoms affects the valence state of metal atoms significantly and plays a key role in the reactivity. Furthermore, the effects of long-range spin coupling and short-range weak metal-metal bonding on OER activity are discussed based on the Cu-N4configuration. By replacing the vicinal metal to regulate the weak metal-metal bonding, the extrapolated overpotential of 0.23 V from the OER volcano curve can be obtained, which is comparable to the noble metal catalysts. This study explores the active sites and influencing factors of OER activity through a series of Co-N-C systems,which guide the design of high-efficiency OER electrocatalysts.
2 Computational methods
A Co and N co-doped graphene system was constructed using a supercell containing a (6 × 6) graphene sheet, in which the Co doped atoms replaced two adjacent C atoms, and the nitrogen atoms were introduced to replace the carbon atoms, directly connecting with the Co dopant atom. Our calculations in this work are done through the Vienna ab initio Simulation Package(VASP) code27,28. The interaction of ions-electrons is described by the projector augmented wave (PAW) method29. The electron exchange and correlation were approximated by Perdew-Burke-Ernzerhof (PBE)30, and the kinetic energy cutoff of the planewave basis was set to 520 eV. In addition, since the weak interaction cannot be accurately described by the standard PBE functional, the DFT-D3 scheme based Grimme empirical atom pair correction is used to describe the long-range van der Waals interaction31,32. The Brillouin zone was sampled using the Γ-centered k-point grid with the sizes of 5 × 5 × 1 and 7 × 7 × 1 kmeshes for structure relaxation and electronic properties calculation, respectively. A 16 Å (1 Å = 0.1 nm) vacuum along z-direction was employed to avoid the interaction between two periodic units. All atoms were fully relaxed until the energies and residual forces on each atom converged to 10-5eV and 0.02 eV·Å-1, respectively, during geometry optimization. The charge transfer between adsorbates and the catalyst was quantitatively described by Bader charge analysis33,34. The climbing image nudged elastic band (CI-NEB) method is used to search for the transition state for the elementary reaction35. With help from the program lobster, the chemical bonding analysis of the metaladsorbate interaction was carried out using the crystal orbital Hamilton population (COHP)36,37.
Following the kinetic model developed by Hansen et al.38, we simulate the polarization curve of Co-N-C. The O electrochemical oxidation steps are listed by the following equations:

where the adsorption sites derived from *O2(aq) represent O2in the electrolyte, specific information is given in Supporting Information.
3 Results and discussion
3.1 Single atom
The identification of the active sites, which establishes a definitive correlation between the atomic structures and catalytic performance, plays a key role in the improvement and new design of single atom catalysts (SACs). Recent advances in experimental studies reported that the SACs can be identified as,mostly, an M-N4configuration by sophisticated techniques. For example, Ban et al. used the electron energy loss spectroscopy(EELS) analysis results of C, N, and Co to prove that more Co-N4was induced during the structural differentiation process17.Meanwhile, Wang et al. calculated the formation energy of the Co-N-C structure and found that with the increase of the N dopant concentration, the formation energy decreased, resulting in a higher possibility of observing the most stable Co-N4in the experiment39. However, the current density of the Co-N-C catalyst in the experiments under low overpotential conditions is relatively low, about 5 to 20 mA·cm-2, which means that a small number of active sites can reach limited values and thus the existence of other configurations as the “real” active sites cannot be ruled out21. Therefore, we collect possible configurations for single atom sites based on reported studies, including Co-N3, Co-N1C3, Co-N2C2, Co-N3C1, Co-N3-v and Co-N4structures (Fig.1a)21,39,40. As proposed by prior studies, a four-step OER mechanism was assumed that proceeds through OH*, O*,OOH*, and O2* (the adsorption site denoted with the asterisk)41.The overpotential corresponding to the OER catalyst is an essential parameter for evaluating the catalytic activity, and it is determined by the free energy of the rate-determining step(RDS)12. Several structures are calculated and compared with the existing data, the consistent results validate our computational models and methods21,40. The detailed calculation results for the elemental steps of all the model structures are given in Table S1 (in Supporting Information).
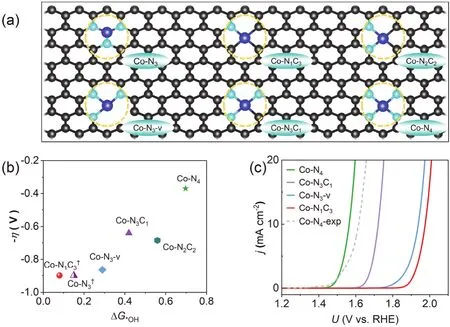
Fig. 1 Evaluation of catalytic activity of single atoms with different structures. (a) Monoatomic structure of cobalt with different coordination structures, where black, light blue and blue represent C, N and Co atoms, respectively (color online). (b) The relationship between the overpotential of different Co-N-C catalysts and ΔG*OH, where † is referenced in Ref. 40. (c) Simulated polarization curves of Co-N4,Co-N4-exp, Co-N3C1, Co-N3-v and Co-N1C3, data of Co-N4-exp is referenced in Ref. 21.
The OH* intermediate, created upon the first electron and proton transfer from water, is expected to initiate the formation of the first chemical bond of O2(O―O), with corresponding free energy differences ΔG*OH. Fig. 1b is obtained according to the overpotential of the Co-N-C catalyst and the ΔG*OH, which was used as a predictive theoretical descriptor of OER activity in previous studies42. In general, the overpotential gradually increases as the bonding strength with *OH increases (ΔG*OHgradually decreases). Such trend corresponds to the increase in the adsorption capacity of the catalyst to certain oxygencontaining intermediates. Based on Sabatier principle, an ideal electrocatalyst needs moderate binding with all the involved intermediates to approach the theoretical value of 1.23 V for each reaction step (4.92 V/4)43. It can be found that Co-N4has the highest ΔG*OHof 0.70 eV, which is closer to the theoretical value compared with other configurations, and its overpotential is the lowest overpotential of 0.37 V among all the investigated configurations. As ΔG*OHdecreases, the overpotentials of Co-N3C1, Co-N3-v, and Co-N1C3are 0.64, 0.87 and 0.90 V,respectively (Fig. S1). The difference in these systems can be essentially explained by the valence state of the central Co atom.Based on Bader charge analysis of Co-N3-v, Co-N3C1, and Co-N4(Table S2), each coordinated N atom gets 0.45e on average,and the C atom gets 0.19e. The Co atom in Co-N3-v loses 0.76e,and the Co atom in Co-N3C1and Co-N4loses 0.80e and 0.89e,respectively, Consistently, it can be found that the adsorption strength of Co-N3C1for oxygen-containing intermediates is slightly stronger than Co-N4. Similarly, for Co-N3-v, the coordination atom number reduction increases the valence state of Co and further increase the adsorption strength of oxygencontaining intermediates on Co-N3-v.
To further understand the influence of Co-N-C coordination structure on reaction overpotential, four reported typical configurations such as Co-N4, Co-N3-v, Co-N3C1and Co-N1C3are selected for further study, and the overpotentials follow the order of Co-N4< Co-N3C1< Co-N3-v < Co-N1C3. From Fig. S1,it can be found that the rate-determining step (RDS) of Co-N4,RDS is OH* oxidized to *O, and the RDS of other catalysts is the oxidation of O* to OOH*12. From the orbital perspective of*OOH formation, an orbital of σ character (highest occupied molecular orbital of OH-) is close to the M-O π* orbital (lowest unoccupied molecular orbital). The attaction of OH-to MO bond leads to the formation of an O―O σ bond in *OOH and the breakage of one M=O π bonds (Fig. S2). In that case, it is requried that the energy levels of M―O π* orbital and OH-σ should be as close as possible to form a new O―O bond.Therefore, the adsorption of *O can be neither too strong(decrease the energy level of M―O π* orbital ) nor too weak(increase the energy level of M―O π* orbital)44. Among these catalysts, the ΔG*OHand ΔGO*of the Co-N1C3system are the lowest. From the adsorption energy diagram (Fig. S3), it is found that the chemical adsorption of the corresponding intermediates is also strong. This leads to nucleophilic OH-attacking the strongly adsorbed *O to form *OOH with a higher energy barrier and overpotential.
As mentioned above, the Co-N4structure shows ΔG*OHof 0.70 eV, which is much less than the theoretical value of 1.23 eV, suggesting the activity can be improved by modulating the charge state of the metal center and the binding energies with the oxygen-containing intermediates. For example, He et al.enhanced the intrinsic activity by adding additional graphitic N doping surrounding the Co-N4moieties, the essence of which is to increase the N doping concentration near the metal center,increase the valence state of the metal center, and promote moderate adsorption of reaction intermediates45. It is also worth noting that the overpotentials in the OER are different at the same N concentration. For instance, Zhang et al. calculated the overpotential of three CoN2C2structures, Co-N2-O (1.81 V), Co-N2-F (1.69 V) and Co-N2-S (1.36 V)39. Thus, the chemical environments of the active metal atoms, including coordination number and local composition, are critical to the activity of SACs.
To give an intuitive demonstration of the OER catalytic performance for Co-N-C, we simulated its polarization curve and compared it with experimental values. At a current density of 10 mA·cm-2, the overpotential of Co-N4is 0.33 V, which was much lower than Co-N3C1(η10= 0.49 V), Co-N3-v (η10= 0.71V)and Co-N1C3(η10= 0.77 V). The activity trend of Co-N-C is consistent with our previous calculation results. It can be observed that the polarization curve of Co-N4structure is closest to the experimental value (η10= 0.39 V)21, thus the dominant SAC sites can be attributed to such a strong coordinated configuration. However, the existence of other configurations,such as Co-N3C1, cannot be ruled out, the gap between the experiment and the theoretical value could be because the active site during the experiment is a mixture of multiple Co singleatom configurations. To sum up, the single-atom structure of Co-N4in Co-N-C has effective coordination, moderate adsorption strength for OER reaction intermediates and the lowest reaction overpotential among all the investigated SAC structures.
3.2 Diatoms and clusters
Due to the lack of precise atomic position control in current synthesis methods, and the catalyst is accompanied by structural reconstruction in the electrocatalytic process, it is possible that M atoms migrate and agglomerate into metal clusters during experimental processes46. For diatoms, we design four structures based on reported experimental results, i.e., Co2-type1, Co2-type2, Co2-type3 and Co2-type4 respectively, as shown in Fig. S4. Considering the three-coordination structure of the Co atom in Co2-type1, we compared it with fourcoordination Co-N4and clusters of atomic states to find the nature of OER overpotential change caused by the structure. In addition, as the distance between active sites decreases, there are interactions between metal centers, and the distance between active centers gradually decreases from Co2-type2 (dCo-Co= 4.98 Å) to Co2-type3 (dCo-Co= 4.08 Å) to Co2-type4 (dCo-Co= 2.25 Å).Therefore, we further study the metal-metal interactions by taking these three diatomic structures for comparison.
Firstly, we further study the oxygen evolution activity of Co-N-C structures containing a metal dimer and four-atom cluster,denoted as Co2-type1 and Co4-type1 (Fig. 2a), respectively. The corresponding OER free energy diagrams are calculated and presented in Fig. 2b. From the free energy diagram, compared with Co-N4, we find that Co2-type1 and Co4-type1 have strong adsorption capacity for the reaction intermediates, leading to considerably large overpotentials of 1.82 V and 0.96 V,respectively, which are much higher than that of Co-N4(0.37 V).It can be noted that free energy of the first step is an energy decreasing process, indicating that the adsorption of *OH can be a thermodynamically favorable process. The high affinity of the Co dimers and clusters towards oxygen intermediates can be attributed to the low oxidation state of the metal atoms. As proved by Bader charge analysis, the Co atom in Co-N4loses 0.89e (0.89e-), which is much more than Co2-type1 (0.67e-) and Co4-type1 (0.10e-). Accordingly, the accumulated charge around the Fermi level of Co2-type1 and Co4-type1 (Fig. S5) can be transferred to oxygen atoms effectively, forming strong chemical bonding between Co atoms and oxygen-containing adsorbates.
The above results also suggest that the Co atoms in dimers and clusters present high reactivity and can be unstable in the current form during OER processes. Alternatively, the free energy changes indicate that metal sites can be adsorbed and stabilized by *OH and *O with accessible potentials. In that case, the “real”catalytic models can be changed to the structures with adsorbed*OH and *O, as the following OER processes is restricted for pristine Co-N-C catalysts due to large overpotentials. Wang et al. recently discovered through DFT calculation and experimental methods that during the ORR reaction, Co2-type1 would spontaneously form *OH as the modified ligand and bind to atomic metal sites, which has higher overall ORR catalytic activity compared with traditional metal surfaces24.Subsequently, the catalytic performance of Co2-type1 and Co4-type1 after pre-adsorption reaction intermediates (such as O,OH, O2) are evaluated. The results show that the oxygen evolution activity is significantly improved by the preadsorption, the calculated overpotentials drops largely from 1.82 V to 0.48 V, 0.40 V and 0.52 V for *OH, *O and *O2preadsorbed Co2-type1 structure. Similarly, the overpotential of Co4-type1-O (Fig. S6) decreases from 0.96 V to 0.40 V with preadsorbed *O species. Meanwhile, the polarization curves of the catalysts after pre-adsorption were simulated (Fig. 2d). At a current density of 10 mA·cm-2, Co2-type1-O (η10= 0.42 V), Co2-type1-OH (η10= 0.42 V) and Co2- type1-O2(η10= 0.46 V) is similar to Co-N4(η10= 0.33 V). It can be noticed that the overpotentials of the cobalt dimers and clusters are significantly improved after pre-adsorption, where the overpotentials of Co2-type1-O are close to the singly occupied Co-N4structures. In addition, the breakage of O=O and desorption of O2are also considered. The calculation results show that O2adsorption can stabilize on the metal dimer and cluster (Fig. S7 and Table S3),the second O2molecule can desorb (binding energy -0.15 eV)without affecting OER activity or causing catalyst oxidation and blocking metal sites after Co2-type1 pre-adsorbs one O2molecule (Fig. S8).
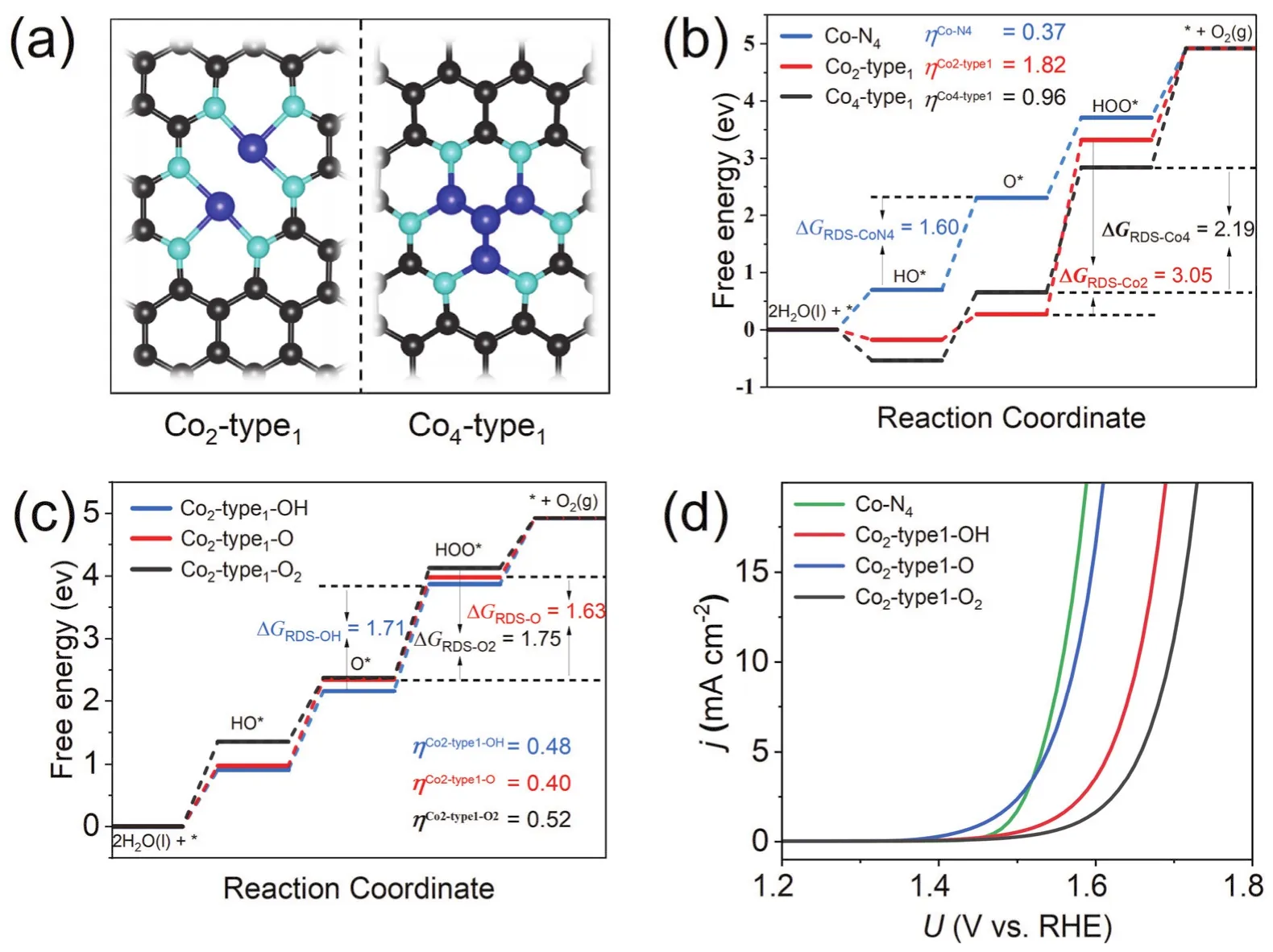
Fig. 2 Evaluation of catalytic activity of diatoms and clusters. (a) Structures for Co2-type1 and Co4-type1. (b) Reaction energy diagram of Co-N4, Co2-type1, and Co4-type1. (c) Reaction energy diagram of Co2-type1 after pre-adsorption of OH, O and O2 (Co2-type1-OH,Co2-type1-O, Co2-type1-O2).(d) Simulated polarization curves of Co-N4, Co2-type1-OH, Co2-type1-O and Co2-type1-O2.
The pre-adsorbed models can also be compared to cobalt(oxygen) hydroxide, which have shown promising OER activity in recent studies47,48. and the high-valence cobalt Co3+(β-CoOOH) is considered to be the active catalytic center of OER process49. The high-valence Co in the (oxygen) hydroxide exhibits the t2g5eg1electronic configuration of the intermediate spin state, and the egorbital directly participates in the σ-bonding between adsorbed species on the surface, the appropriate egorbital filling is beneficial to the oxygen-containing intermediates in the process of oxygen electrocatalysis (such as OH*, O* and OOH*) interact50. Therefore, the catalytic activity of Co-based catalysts is highly depending on the high coordination number and the oxidation states of the metal sites,rather than composition forms (single atom or metal clusters).
3.3 Indirect metal-metal interactions
According to the above results, the high coordination is a prerequisite for the OER process and Co-N4presents the moderate adsorption energies for each reaction intermediates.Due to the uneven distribution of reaction sites and different metal doping concentration applied in the experiments, when the distance between active centers decreases, there can be interactions between two adjacent reaction sites, such as the long-range spin coupling effects between metal centers and weak metal-metal bonding51,52. Considering the spin nature of electrons in the OER mechanism, the adsorption of OER reaction intermediates at the metal center can be strongly regulated by the spin state, which has been extensively studied53.However, the effect of inter-site spin coupling is less discussed,thus several reported configurations with different Co-Co distances are considered to study the long-range interactions.Firstly, Co2-type2 and Co2-type3 were established to study the influence of spin coupling between adjacent metal centers on catalytic performance (Fig. 3a), the distances between the two Co atoms are 4.96 Å and 4.08 Å, respectively. As shown in Fig.3a, the energy differences of Co2-type2 and Co2-type3 between ferrimagnetic and anti-ferrimagnetic (energy favorable state) are 60 meV and 156 meV, respectively, indicating that the spin coupling effect becomes more pronounced as the distance decreases. Comparing the change in reaction free energy (Fig.3b), the overpotential of Co2-type2 with the antiferromagnetic state is similar to that of Co-N4, 0.39 V and 0.37 V, respectively.On the contrary, the overpotential of Co2-type3 (ηOER= 0.48 V)is higher than that of Co-N4(ηOER= 0.37 V) and the RDS of Co2-type3 (oxidizes O* to OOH*) is different from Co-N4(oxidizes OH* to O*).
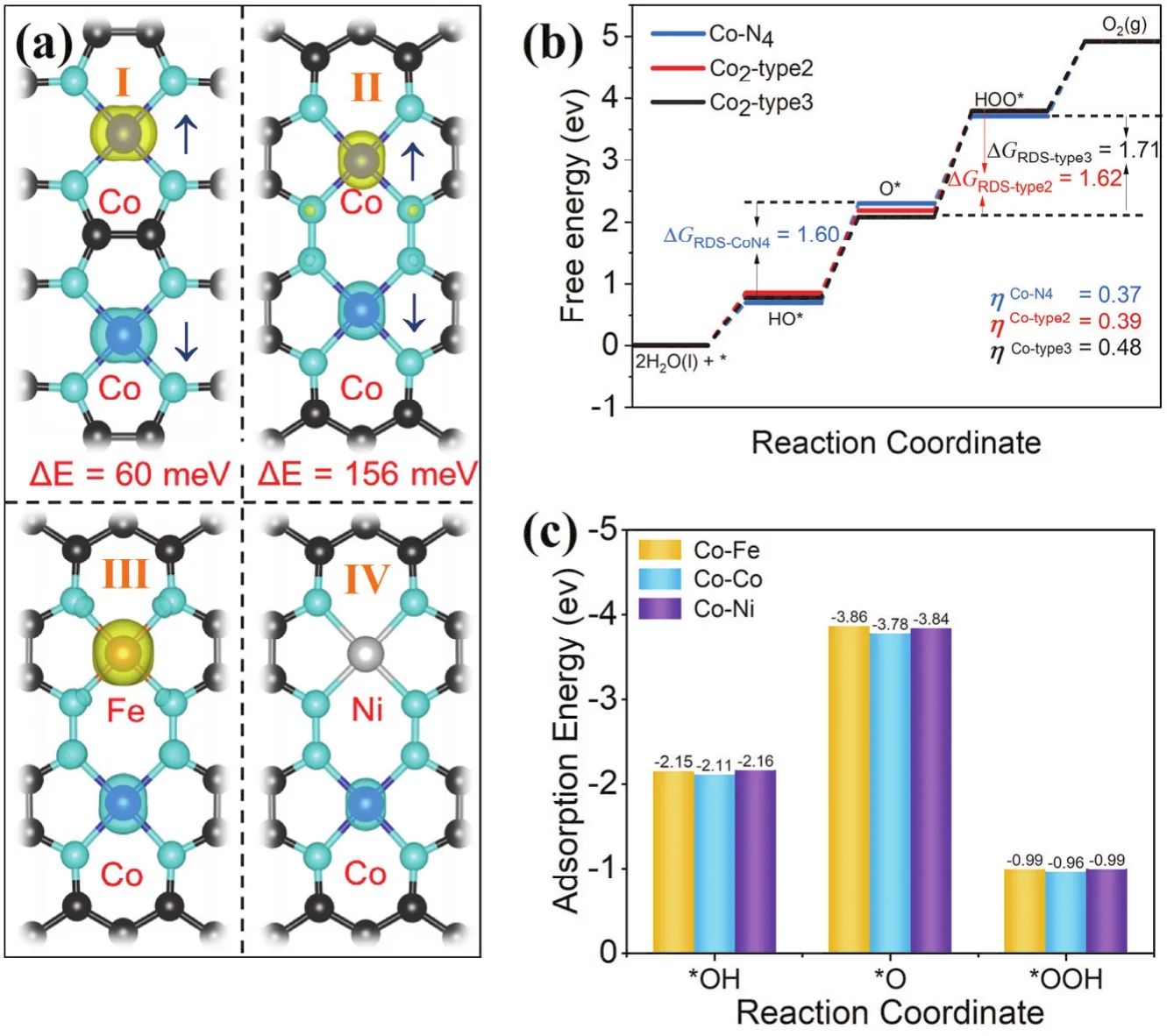
Fig. 3 Identification of the indirect inter-site interaction. (a) Spin density of Co2-type2 (Ⅰ), Co2-type3 (Ⅱ ), CoFe -type3 (Ⅲ ) and CoNi- type3 (Ⅳ).The yellow or blue color indicates positive and negative spin density. The isosurface is plotted with 0.003 eV·Å-3. (b) Reaction energy diagram of Co-N4 (blue), Co2-type2 (red) and Co2-type3 (black). (c) Adsorption energies of oxygen-containing intermediates (*OH, *O, *OOH) on CoFe-type3, Co2-type3 and CoNi-type3, respectively. Color online.
To further study the effect of spin coupling between active centers on the activity of OER catalyst, we replaced one of the two Co atoms with the strong magnetic Fe and weak magnetic Ni. The structure and spin density are shown in Fig. 3a, and the spin configurations are presented in Fig. S9. Then, the OER process was calculated at the Co site for comparison (Fig. S10).The free energy diagrams of CoFe-type3, Co2-type3, and CoNitype3 show that the overpotential and RDS of OER have no apparent change after the replacement. Compared with the adsorption energy of the reaction intermediates of Co-Fe, Co-Co, Co-Ni in Fig. 3d, there is no significant change, which shows that the spin coupling between active centers has a minor effect on the activity of the reaction centers. To clarify the difference between Co2-type3 and Co-N4, Bader charge analysis is performed (Table S1) and the results show that the charge of the N atom located between the two Co centers is 0.45e-lower than the surrounding N atoms of Co-N4, indicating the local environment is changed. Therefore, the difference in RDS and OER performance between Co2-type3 and Co-N4can be attributed to the influence of the coordination environment,rather than the spin coupling between the two reaction centers.
As the distance between two Co-N4structures approaches and forms a closely connected structure, i.e., Co2-type4 (Fig. 4a), the short distance can induce metal-metal bonding between two adjacent active centers thus have influence on the catalytic performance. The bonding is expected to be weak as the high coordination nature of the metal sites. As from the electron localization functions (ELF) contour analysis (Fig. 4a, bulk Co is shown in Fig. S11), it can be regarded that there is a weak bond formation between the two Co atoms and the electrons (density)are mainly localized on the surrounding N atoms. The free energy diagram of Co2-type4 is calculated and compared with Co-N4, as seen in Fig. 4b. The overpotential of Co2-type4(yellow) is 0.36 V, which is the same as Co-N4(black), 0.37 V.However, the free energy change of Co2-type4 on each step is different from Co-N4. To further determine the effect of the weak metal-metal bonding, a Co atom is replaced in Co2-type4 with Mn, Fe, Ni, and Cu, and the free energy diagram of the oxygen evolution reaction at the same Co site is calculated (Fig. 4b). The result shows that when the adjacent metal changes, the change of the RDS for the type4 configuration mainly depends on the species O*. Moreover, ΔGO*changes obviously in a linear way and the adsorption strength of O* decreases in the order of Mn >Fe > Co > Ni > Cu (Fig. S12). In addition, we considered the influence of the pre-adsorption intermediates at the ortho metal site on the performance of the Co site, and the results showed thatortho metals of type4 configuration can adsorb oxygencontaining intermediates, but the effect on the OER performance of the Co metal atoms is negligible (Fig. S13).

Fig. 4 Identification of the weak metal-metal interaction. (a) Structures for Co2-type4 and the electron localization functions (ELF) contour.(b) Reaction energy diagram of Co-N4, CoMn-type4, CoFe-type4, Co2-type4, CoNi-type4, and CoCu-type4. (c) The scaling relationship between ΔG*OOH and ΔG*OH. (d) The scaling relationship between ηOER versus ΔGO* - ΔG*OH. The theoretical volcano plot derived from the data is displayed in black. Color online.
To further understand the change rule of O binding strength on Co atom, we carried out Crystal Orbital Hamilton Population(COHP) analysis. The projected COHP (pCOHP) and the integrated COHP (ICOHP) for the Co―O bond in O-Co2-type4,O-CoNi-type4, and O-CoCu-type4 systems with consistent adsorption sites are calculated (Fig. S14). In the figure -pCOHP is plotted so that the population on the right of the x axis indicates the bonding state and those on the left indicate the antibonding state. The ICOHP is obtained by integrating pCOHP with Ef,which is a measure of bonding strength. The more negative the ICOHP value is, the stronger the Co―O bond will be. From the COHP analysis, we note that from Co2-type4 to CoCu-type4, the part of the antibonding state above the Fermi level gradually increases, and the antibonding state is less filled, which enhances the adsorption of O*. The ICOHP value of Co2-type4 is -0.76 eV, and its Co―O bond strength is the weakest (Fig. S14).Followed by CoNi-type4 (-0.91 eV) and CoCu-type4 (-0.96 eV), the Co-O bond is strengthened in turn, which is consistent with the above analysis. Therefore, the weak metal-metal bonding can influence the OER activity and the adsorption energy of O* can be regulated to optimize the OER performance,especially the hetero-metals.
According to previous findings, the activity of OER is strongly related to ΔGO*; thus, it can be used as an adequate descriptor to predict the overall OER reactivity, which involves multiple intermediates. Subsequently, the minimum overpotential can be predicted by establishing the scaling relationship between the adsorption free energies of different adsorption species. As shown in Fig. 4c, the linear relationship between ΔGOOH*and ΔG*OHis ΔGOOH*= 0.69ΔGO*+ 3.32, and the coefficient of determination (R2) is 0.945, indicating that a fine linear relationship is obtained. This correlation shows that the catalytic activity of OER can be described by the free energy of adsorption of one or two intermediates. By plotting the relationship between the oxygen evolution overpotential of different Co-N-C structures and ΔGO*- ΔG*OH, it is found that the binding strength of the adsorbed species, as well as the overpotential, is in a volcanic shape as shown in Fig. 4d. Besides,it is worth mentioning that the RDS of the system on the left side of the x axis 1.47 eV is *O oxidized to *OOH, and the RDS of the system on the right of 1.47 eV is the oxidation of *OH to *O.The reason is that the system on the left has strong adsorption on oxygen-containing intermediates, and the stronger adsorption of OH also activates OH, which reduces the energy barrier in the step of *OH oxidation to *O. However, the strong adsorption of O* on metal sites leads to the nucleophilic ―OH attacking *O,forming *OOH with a high energy barrier (―OH combines with O―M to form O―O―σ bond and break one of the M―O π bonds at the same time), which limits the catalytic process44.The higher ΔGO*value on the right side of 1.47 V on the x axis in Fig. 4d indicates that the adsorption capacity of *OH on the catalyst is weak, which is not conducive to OH activation,resulting in a higher energy barrier for *OH dissociation from H+to form O*. For all the calculated systems, CoNi-type4 lies on the top of the volcano curve and present the best OER performance, consistent with the free energy calculation results for all the elementary steps. Besides, the volcano curve shows that the catalytic activity of OER reaches its peak when ΔGO*is close to 1.47 eV, and the minimum ηOERis 0.23 V, which exceeds the limit overpotential of 0.37 V in the volcano curve of previous OER catalysts, breaking the OOH to OH ratio relationship54,55.
4 Conclusion
In this work, a series of Co-N-C catalysts with different configurations (single atom, diatoms, and cluster) have been established to determine the origin of the OER activity. Based on in-depth analyses including overpotential, adsorption energy,PDOS, Bader charge and COHP, the results demonstrate that the high coordination of metal sites with a high valence state is required to ensure the moderate adsorption strength of the reaction intermediates, which makes Co-N4as the best catalyst among all the established models. Metal dimers and clusters need to be further re-constructed by the interaction with catalyst supports (interface engineering) and the adsorption of solvation ions and reaction intermediates. Besides, the binding strength of the reaction intermediates can be also modulated by the indirect change of the coordination environment, which endows the change of the micro-environment of the catalysts to be one of the common ways to promote the reaction activity. Moreover,hetero-atom doping with weak metal-metal interactions is proven to be another way to promote reaction activity. In a combination of these strategies, a volcano curve is established by associating the overpotential of different Co-N-C structures with ΔGO*, and an ultralow overpotential of 0.23 V is obtained,which could break the theoretical optimum value (0.37 V). This study explores the structure-activity relationship on OER through the refined Co-N-C structures, which bridges the gap between the experiments and theoretical calculations and thus provides a theoretical basis for the rational design of highefficiency OER electrocatalysts.
Acknowledgements: The authors thank the computational resources from the Big Data Center of Southeast University and National Supercomputing Center of Tianjin.
Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn.
