SLC25A20 基因c.199-10T>G 纯合致肉碱酰基转移酶缺乏症1 例及家系基因报告
2023-01-09邓朝晖潘嘉浩吴国豪黄宇戈
邓朝晖,潘嘉浩,吴国豪,黄宇戈
(1.广东省吴川市妇幼保健院儿科,广东吴川 524500;2.广东省吴川市妇幼保健院新生儿科,广东吴川 524500;3.广东医科大学附属医院儿童医学中心,广东湛江 524000)
肉碱酰基转移酶缺乏症(CACTD,OMIM 212138)为常染色体隐性罕见遗传病,是由于肉碱-酰基肉碱移位酶功能缺陷引起,导致长链酰基肉碱不能进入线粒体内膜参与β 氧化,是线粒体脂肪酸β 氧化代谢异常的一种类型[1]。CACTD 大多在新生儿期起病,进展快、预后差、病死率高。目前国内有关CACTD 报道极少,现分享1 例经基因检测为SLC25A20基因c.199-10T>G纯合致CACTD 及其家系基因报告,旨在提高临床医师对该疾病的认识。
1 病例资料
患儿,女,第2 胎第2 产,胎龄38+3周,于2021 年11 月5 日2 点50 分经阴道顺产分娩,出生后一般情况尚可,1、5、10 min 阿氏评分均为10 分,出生体质量为2.67 kg,约当天6 点11 分发现患儿全身肤色紫绀伴意识丧失,呼吸、心跳停止,刺激未见反应。家族史:否认近亲结合,其余无特殊。入院查体:肛温35.6℃,心率0,呼吸0,血压测不出,足月儿外貌,无反应,全身肤色紫绀,前囟饱满,颈软,双侧瞳孔等大、等圆,约4 mm 大小,对光反射消失,唇青紫,腹平软,肝脾肋下未及,肠鸣音消失,四肢肢端青紫,毛细血管充盈实验(CRT)约8 s,四肢肌力、肌张力消失,各原始反射消失。
辅助检查:入院血糖1.4 mmol/L;血气提示pH 7.16,PO2248 mmHg,PCO244.8 mmHg,Lac 8.8 mmol/L,HCO3-15.8 mmol/L,K+6.53 mmol/L,Na+133 mmol/L,Cl-103 mmol/L,Ca+0.92 mmol/L;血常规示:WBC 21.46×109/L,RBC 4.77×1012/L,HGB 174 g/L,PLT 405×109/L,CRP 8.0 mg/L;生化:AST 211.40 U/L,ALT 40.50 U/L,CK 9 390.40 U/L,CK-MB 273.00 U/L,HBDH 657.20 U/L,LDH 1 036.10 U/L。心脏彩超:卵圆孔未闭、动脉导管未闭、心律失常。头颅超声:脑实质弥漫性改变,考虑新生儿缺血缺氧性脑病。胸片:双肺野见小斑片絮状阴影。
治疗经过:入院立即行气管插管接呼吸囊(纯氧)正压通气,持续胸外按压,多次予1/10 000 肾上腺素静推,生理盐水扩容、碳酸氢钠纠酸、10%葡萄糖静推等处理后患儿心率、呼吸恢复,住院期间患儿多次心率、呼吸下降,伴反复低血糖、高钾血症、低钠血症等电解质紊乱,予维持心率、呼吸、血糖、电解质平衡,考虑患儿可能存在遗传代谢病,予葡萄糖6 mg/(kg·min)持续输注补给以提供能量供应,及左卡尼丁(100 mg/kg)生物碱辅助治疗后病情仍继续加重,出现抽搐等脑损伤以及多器官功能衰竭,预后极差,完善血尿串联质谱筛查、全外显子高通量检测及线粒体体高通量检测,家长放弃治疗,患儿出院后死亡。
2021 年11 月8 日血遗传代谢病串联质谱分析检测报告:Cit、GAA 升高,C0、C2、C3、C4、C4-OH降低及相关比值异常(表1)。2021 年1 月22 日全外显子组测序检测报告受检者(患儿)携带SLC25A20(NM_000387.5)基因的纯合致病,其突变位点为c.199-10T>G,判断为致病变异,且体内外功能实验已明确该变异会导致基因功能受损,是ESP 数据库、千人数据库、EXAC 数据库中正常对照人群中未发现的变异。其父母经过Sanger 验证检测携带SLC25A20基因c.199-10T>G 杂合突变(图1)。
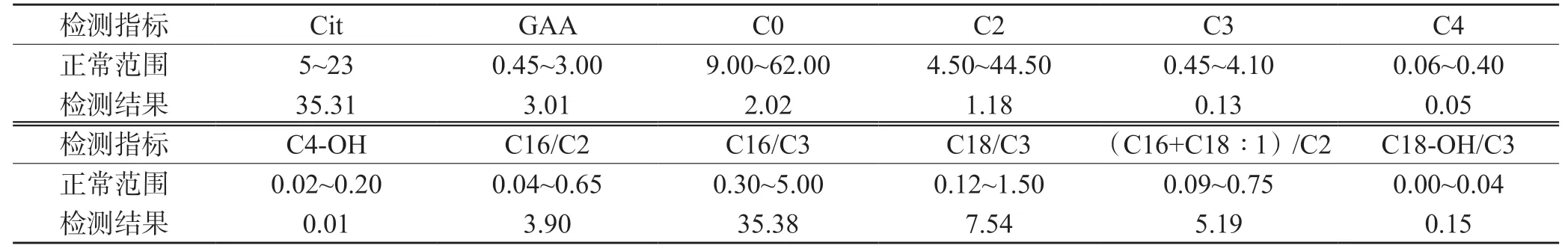
表1 患儿血遗传代谢病串联质谱分析结果
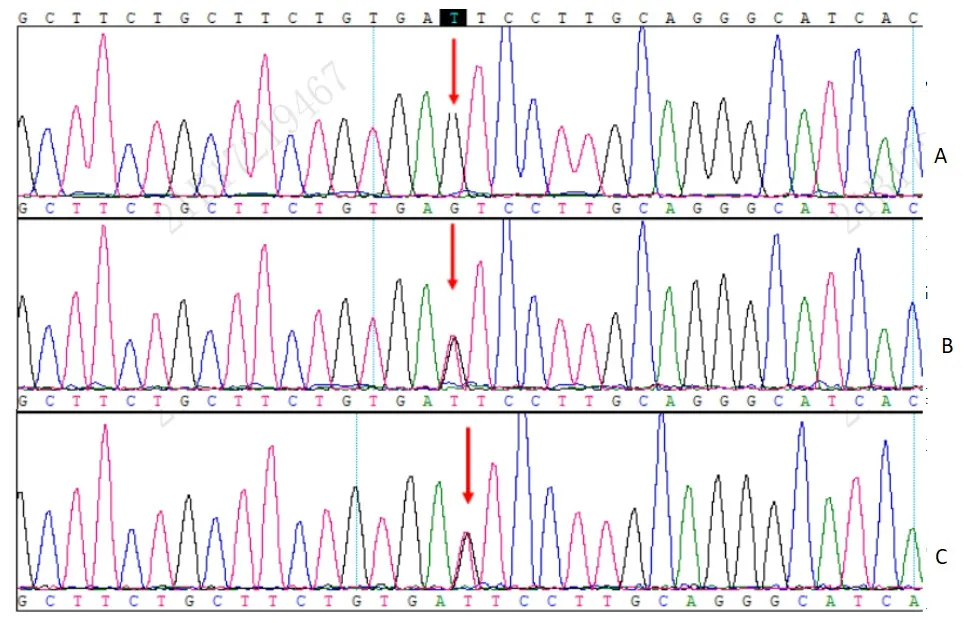
图1 患儿及家系Sanger 测序验证图
2 讨论
CACTD 是一种罕见且致死性高的常染色体隐性先天性疾病[2]。CACT 蛋白由SLC25A20基因编码,该基因位于染色体3p2l.31。本案例通过全外显子组测序检测SLC25A20基因9 个外显子编码区及外显子-内含子交界处,发现c.199-10T>G 纯合突变,为已知致病突变,且是报道当中最常见的SLC25A20基因突变。根据患儿旳检测结果,对其父母SLC25A20基因3 号外显子编码区及外显子-内含子交界处进行检测,均发现c.199-10T>G 杂合突变,符合常染色体隐性遗传病遗传规律。Hsu 等[3]研究发现c.199-10T>G 突变型的患儿结局差、死亡率高,可能是由于c.199-10T>G 突变对mRNA 产物的影响,使得CACT 酶蛋白被截短,导致转位酶无活性。同时因不同变异导致肉碱-酰基肉碱移位酶活性降低程度不同,使其临床表型轻重不一,严重者残余CACT 酶活性不及正常水平的1%,出生后不久即死亡,轻型者具有大约5%残余酶活性,经早期治疗后可存活至青春期以后[4-5]。
CACTD 的病理机制是CACT 缺乏而导致长链脂肪酸不能进行线粒体β 氧化和酮体生成,由于CACTD基因在心脏、肝脏和骨骼肌表达广泛,使得长链酰基肉碱易堆积于此类组织,从而导致大多病例在新生儿期发病就出现肝酶和肌酸激酶增高,且多数病例合并心律失常和惊厥,又因肝糖原耗竭、糖原再生受损、酮体生成障碍导致低血糖[1,6-7]。因此CACTD 主要临床表现为低酮性低血糖、心律失常、昏迷、抽搐,甚至猝死;生化检查结果大多以低血糖、高血氨、高血钾、肌酸激酶升高;血串联质谱提示长链酰基肉碱异常升高为突出表现。
CACTD 急性期治疗包括:静脉输注葡萄糖,降血氨,频繁喂养,低脂饮食,限制长链脂肪酸的摄入,可用中链甘油三酯代替。CACTD 患儿肉碱的补充仍存在争议,肉碱的补充剂量也不一致,范围一般为30~150 mg/(kg·d)[8]。
新生儿CACTD 起病早、病情急重,病死率高,临床表现缺乏特异性。当儿科医师遇到新生儿早期不明原因低血糖伴高血钾、心律失常及肌酶显著升高等,疑脂肪酸氧化代谢障碍类疾病时,可在确诊前进行相应的治疗,如静脉输注葡萄糖溶液保证高热量供给,适当补充左卡尼汀等。同时应尽早行质谱血、尿代谢物检测,基因检测是目前明确诊断的最可靠方法。
