团簇NiCo2S4的磁学性能
2023-01-03曾鑫渔方志刚吕孟娜
曾鑫渔,方志刚,吕孟娜,许 友
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
超级电容器作为替代储能装置应用于轨道交通、可再生能源、电力系统等领域,因其具有高功率密度、快速充电能力、优异的循环寿命、高库仑效率和操作安全等优点,近年引起人们的广泛兴趣[1]。但其相对有限的能量密度[2]使其无法在日常生活中广泛应用,而该性能在很大程度上取决于电极材料的规格、容量、速率能力、功率密度[3],因此目前的研究工作主要集中在提高超级电容器高性能电极材料的开发上。特别是三元镍钴硫化物(NiCo2S4),因其理论容量高、成本低、自然丰度高、电导率高、化学氧化还原比其单组分氧化物更丰富[4],被认为是超级电容器最有前途的电极材料之一[5]。电极材料的结构、组成和形貌对其电化学性能具有显著的影响[6],目前已有的科研成果大多是通过改变其原子比例和材料结构来对其电化学方面的性质进行优化探究。如Hui等[7]采用环氧树脂填充、炭化和水热生长NiCo2S4纳米棒的方法,制备出具有更优异电化学储能性能的电极材料;Wang等[8]与Wang等[9]均是通过改变硫空位量来影响材料性能,寻找出具有最佳析氢活性和电化学性质的硫空穴位量。Li等[10]通过调节水和乙二醇的比例来改变NiCo2S4的结构,从而增强其电化学性质和循环稳定性。通过上述实验虽能够表现出材料的宏观性质,却无法体现其微观结构,而微观结构是其性能表征的根本原因,因此对材料微观结构的研究具有着重要意义[11]。考虑到该体系中Co、Ni是具有铁磁性的金属,本文将从成单电子的磁矩及轨道态密度[12]等角度研究该材料的电子结构和磁学性质,以期使其在生产生活中得到更为广泛的应用。
1 计算方法
利用拓扑学原理[13]设计出的近50种初始构型,采用密度泛函理论[14]中的B3LYP杂化和Lanl2dz赝势基组对初始构型进行结构优化和频率等参数运算[15],其中对Co、Ni、S 3个原子使用ECP加double zeta优化[16],并在S原子上添加极化函数ξS,d=0. 55[17]。采用Gaussian09程序在启天M4390计算机上完成上述计算过程。
2 结果及讨论
2.1 团簇NiCo2S4的优化构型
单重态为闭壳层,根据泡利不相容原理,在同一轨道上的两个电子的自旋方向要彼此相反,因单重态分子的电子是自旋成对的,净自旋为零,所以单重态的物质具有抗磁性。因此在探究团簇磁学性质时仅考虑三重态的情况。团簇NiCo2S4的6种优化构型如图1所示,以能量最低的构型1(3)为参考零点(设其能量值为0 kJ/mol), 将其依据能量由低到高排序,其中编号右上角括号内数字表示重态,编号后括号内的数字表示相对能量。这6种优化构型的空间结构有五棱双锥型和单帽四棱双锥型,其中又以五棱双锥型结构居多。空间结构为五棱双锥型构型的分别是1(3)、4(3)、5(3)、6(3),单帽四棱双锥型构型的分别是2(3)、3(3)。构型5(3)和构型6(3)结构的相似度极高,从原子排布上来看,仅是相同原子排列组合方式不同。

图1 团簇NiCo2S4优化构型图Fig.1 Optimal configurations of cluster NiCo2S4
2.2 轨道成单电子数
由分子电流假说可知,磁性产生的根本原因是粒子的自旋运动产生磁场,根据影响强度的大小分别为成单电子的自旋运动、轨域运动以及核自旋运动,本文主要从影响因素入手,用各轨道的成单电子数目来判别团簇NiCo2S4构型磁性强弱。根据电子自旋方向将电子分为自旋方向向上的α电子(数值上体现为正值)和方向向下的β电子(数值上体现为负值)。根据泡利不相容原理,在同一轨道内这两者运动产生的磁效应会相互抵消,剩余未被抵消的电子称为成单电子,因此,成单电子数对物质磁学性质的研究具有重要意义。将团簇NiCo2S4各个构型的轨道成单电子列于表1,表中正负表示方向,绝对值表示成单电子数,sum是各轨道成单电子数目的总和,即前3列数据取绝对值后的总和。根据上文可知,在假设各电子所产生的磁效应一致时,成单电子数绝对值越大对应产生的磁效应就越强。对于团簇而言,团簇整体磁性就是团簇内各成单电子对外界产生的磁效应总和。对应表1中sum一列数据可知,构型2(3)的磁性最强,其余构型的磁性强度相近;在团簇各构型中,成单电子数最多的轨道是d轨道,且为α成单电子,最少的是s轨道,除构型1(3)和构型4(3)外其余构型均是α成单电子,说明对团簇NiCo2S4磁性强度贡献最大的是d轨道,最小的是s轨道,同时结合整体数据可知,α成单电子是团簇整体磁性强度的主要贡献者。

表1 团簇NiCo2S4各构型的轨道成单电子Table 1 Orbital single electrons for each configuration of cluster NiCo2S4
2.3 自旋布居数
自旋布居数反映出某一区域内的成单电子分布情况,将各原子各轨道自旋布居数列于表2,这也是在2.2小节基础上更深入的分析团簇NiCo2S4磁性的影响因素。

表2 团簇NiCo2S4各构型中各原子各轨道自旋布居数Table 2 Number of spin population of each atomic orbital for different configurations of cluster NiCo2S4
结合表1可知,团簇NiCo2S4整体应是受α成单电子影响较大的,因为β成单电子产生的磁性会与α成单电子所产生的相抵消,因此,后续分析过程中出现的受β成单电子的影响将被认为对团簇整体磁性强度起抑制作用。分析表2可知,金属原子在大部分构型中的s轨道是负值,而S原子是正值,金属原子的s轨道自旋布居数主要受β成单电子的影响,S原子主要受α成单电子的影响;Ni、S原子在大部分构型中的p轨道是正值,Co原子是负值,即Ni、S原子p轨道自旋布居数主要受α成单电子的影响,Co原子主要受β成单电子的影响;Ni原子的d轨道大部分构型是正值,其余原子正负情况不等,即Ni原子d轨道主要受α成单电子影响,Co、S原子既受α成单电子影响又受β成单电子影响。再依据2.2小节关于团簇NiCo2S4整体应是受α成单电子影响较大的结论,可得Ni原子对团簇整体的磁性强度贡献最大。轨道之间相比,金属原子的d轨道成单电子数绝对值明显大于另外两者,S原子则是p轨道最大,又因这两部分都是正值,因此,S原子是影响团簇NiCo2S4p轨道成单电子数的因素,金属原子则是影响d轨道的主导因素。特别是构型2(3)的Ni、Co1、Co2、S1、S2原子,除Co2-d、S1-d、S2-d轨道外所有轨道均是负值,S3、S4均是正值,结合表1,表明s轨道成单电子数的主要贡献者是S原子,p、d轨道主要是金属原子,且对d轨道成单电子数实际起作用的是S3和S4两个原子,综上所述说明空间位置对其轨道电子排布有一定影响。
2. 4 自旋磁矩
由前文可知,磁矩可由电子和核自旋产生,但核自旋产生的磁矩远小于电子,故忽略不计,因此自旋磁矩仅考虑电子自旋磁矩。目前常用自旋布居数n(成单电子数)与玻尔磁子μB的乘积表示电子自旋磁矩M。μB的国际标准定义为
式中:e为电荷量;h为普朗克常数;me为电子质量。计算得出μB=9. 274×10-24J/T。这里可以看出μB是常数,即Mn,因此,可以用原子的自旋布居数来讨论磁矩。将团簇NiCo2S4各构型中各种原子产生的自旋磁矩与波尔磁子的比值绘制成图2。由图2可以看出,金属原子中除构型2(3)的金属原子和构型1(3)的Co原子外,其余均是正值,S原子都是正值,说明团簇NiCo2S4中磁矩M由α成单电子自旋运动产生的概率更大。根据图2可以明显对比,除构型2(3)外其余构型间各原子的磁矩大小变化并不大,说明各构型间的差异对原子形成团簇后的成单电子排布影响小。结合2.3小节分析可知,构型2(3)的Ni、Co原子自旋磁矩受β成单电子影响,S原子则是受α成单电子影响,且三者的绝对值相比于其他构型的原子要大得多,尤其是S原子,说明构型2(3)的磁学性能所受成单电子的影响情况与大多数构型不同,且表现优异。另外,构型1(3)的Ni、S原子成单电子数在剩余构型中最多,Co原子因受β成单电子影响,对整体表现为受α成单电子影响的构型起抑制作用,综上来看,热力学稳定性最好的构型1(3)在磁学性能上表现一般。
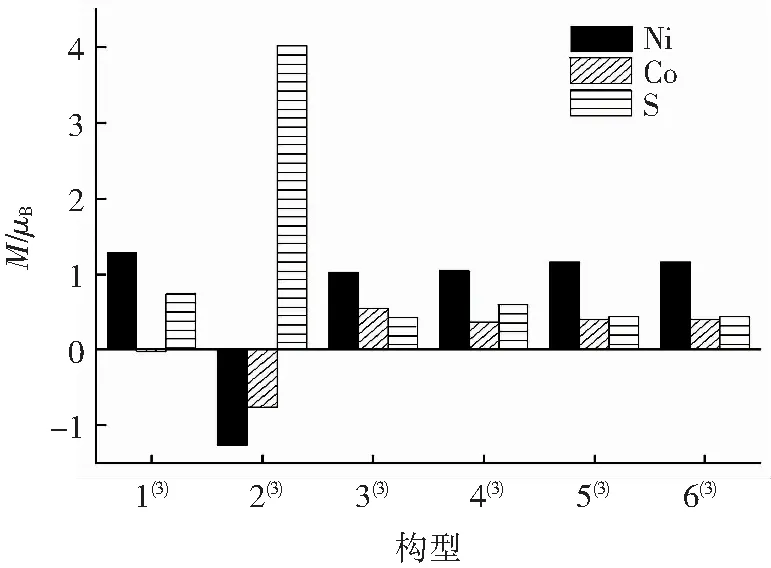
图2 团簇NiCo2S4各构型原子磁矩变化情况Fig.2 Variety of atomic magnetic moment of each configuration of cluster NiCo2S4
2.5 原子电子自旋密度差图
原子电子自旋密度差表示在各原子上自旋向上的α电子和自旋向下β电子的分布密度差值,故其图示可以直接显现原子上的净电子自旋密度分布情况,由于净电子自旋密度大小可以决定磁性强弱,故该图还能够间接表现团簇各构型内各原子的磁性强弱。因此,本文绘制了原子电子自旋密度差图于图3,图中绿色阴影表示电子自旋密度差值为α电子,即该部分磁性由α电子提供,而蓝色阴影则表示为β电子。团簇NiCo2S4中含有3个金属原子,从图3中可以明显看出,所有构型的3个金属原子均有两个原子的净电子密度分布为α电子,另外一个为β电子,其中Ni原子在除构型2(3)的其余构型中均分布净α电子,即可以得出净α电子对金属原子的磁性贡献较大;4个S原子上几乎没有分布净电子,仅少数构型上个别S原子分布少量净电子,如构型1(3)和构型4(3)中一个S原子上分布着少量净α电子,构型2(3)为少量β电子。因此团簇NiCo2S4整体磁性强度由金属原子提供,S原子对整体磁性贡献并不大。整体来看,图3中绿色阴影面积大于蓝色阴影面积,说明团簇NiCo2S4的磁性大多由净α电子提供。

图3 团簇NiCo2S4各构型中原子电子自旋密度差Fig.3 Atomic electron spin density difference in each configuration of cluster NiCo2S4
2. 6 轨道态密度图
轨道态密度(DOS)表示自由电子在相应能量区域间隔的分布情况。将团簇NiCo2S4各个构型的轨道态密度图绘于图4,每个构型有3个小分图,从左至右分别表示对应构型s、p、d轨道的态密度图,图中实线表示自旋向上的α电子,虚线表示自旋向下的β电子,X轴上下曲线积分之和表示轨道成单电子数,即为上下曲线与X轴所围成的面积差值。α电子和β电子因自旋在相同能量片段产生的磁矩会相互抵消,因此反映在轨道态密度图时,图像沿X轴对称性越高,α电子和β电子数量越接近,表示对应轨道的成单电子数越少,那么最终产生的磁性强度就会越弱,反之则越高。团簇NiCo2S4所有构型s轨道的低能量区域(-15~-5 V)对称性相对较差,但上下曲线所围成面积差值不大,两种电子的数目也相差不大;构型2(3)、3(3)和4(3)p轨道的低能量区域(-15~0 V)的对称性不高,且X轴上方曲线面积大于下方曲线面积,因而这些构型的p轨道的成单电子为自旋向上的α电子;所有构型的d轨道在低能量区域(-15~0 V)和高能量区域(25~40 V)中对称性相对不高,X轴上方曲线的总面积高于下方,可得该轨道的成单电子为自旋向上的α电子。综上分析可得,团簇NiCo2S4在低能量区的成单电子数更多,对整体磁性强度的贡献更大,且大多为α成单电子。从左向右对比s、p、d轨道可知,各个构型的s、p轨道态密度图对称性相对更高,表明两轨道的成单电子数较少,s、p轨道对团簇NiCo2S4整体的磁性强度贡献较小,d轨道对整体的磁性强度贡献最大。

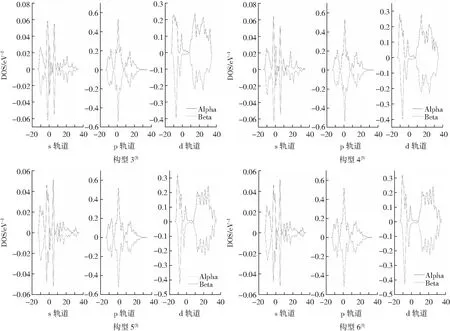
图4 团簇NiCo2S4各构型s、p、d轨道态密度分布Fig.4 Density distribution of states of s, p, and d orbitals of different configurations of NiCo2S4
在2. 2小节部分已分析构型2(3)相比其他构型剩余更多成单电子数,而使其电子自旋磁矩产生的磁性更强。且在态密度图中:s轨道低能量区(-15~0 V)、p轨道的低能量区(0~10 V)以及d轨道的低能量区(-15~0 V)、高能量区(15~40 V)的对称性不高。综上所述,构型2(3)比其他构型拥有更为优异的磁学性能。
3 结 论
本文分别从轨道成单电子数、自旋布居数、磁矩、自旋密度差、轨道态密度图5个方面对团簇NiCo2S4在三重态下的6种优化构型进行了分析,并得出以下结论。
1)团簇NiCo2S4的6种构型中,构型2(3)磁学性能最优异,而热力学稳定性最好的1(3)在磁学性能上表现一般,其余构型磁性性能相近。
2)结合成单电子数、磁矩和原子电子自旋密度差,对构型磁性贡献最大的是d轨道,最小的是s轨道,且Ni原子和α成单电子是团簇整体磁性强度的主要贡献者。其中,s轨道成单电子数的主要贡献者是S原子,p、d轨道主要是金属原子;Ni原子d轨道主要受α成单电子的影响,Co、S原子既受α成单电子的影响又受β成单电子的影响。
3)从轨道态密度图的对称度可知,团簇NiCo2S4在低能量区的成单电子数相对较多,对整体磁性强度的贡献更大,且大多为α成单电子。进一步对比整体s、p、d轨道可知,各个构型的s、p轨道态密度图对称性相对更高,表明两轨道的成单电子数较少,s、p轨道对团簇NiCo2S4整体的磁性强度贡献较小,d轨道对整体的磁性强度贡献最大。
