Role of Macrophage-derived Osteoclasts in Vascular Calcification*
2022-12-22WANGZhongQunZHANGLiLiZHAOYunYunLiHuaYUANWei
WANG Zhong-Qun,ZHANG Li-Li,ZHAO Yun-Yun,LⅠLi-Hua,YUAN Wei*
(1)Department of Cardiology,Affiliated Hospital of Jiangsu University,Zhenjiang 212001,China;2)Department of Pathology,Affiliated Hospital of Jiangsu University,Zhenjiang 212001,China;3)Guangxi Key Laboratory of Diabetic Systems Medicine,The Second Affiliated Hospital of Guilin Medical University,Guilin 541100,China)
Abstract Vascular calcification is a cell-mediated active biological process, similar to bone remodeling, and plays an important role in the occurrence and evolution of acute and chronic cardiovascular and cerebrovascular events.Ⅰn recent years,the research on the mechanism and prevention of vascular calcification has gradually attracted the attention of scholars, but unfortunately, precise molecular and cellular targeted therapy for clinical application is rare. Previous studies have shown the presence of the osteoblast phenotype and dysfunctional osteoclasts in atherosclerotic plaques of diabetes.The imbalance of osteoblasts and osteoclasts may be the key step in calcification development in atherosclerotic plaques. Ⅰt is known that macrophage-derived osteoclasts are the only cells with bone resorption activity and have the potential to reverse calcification.Therefore,exploring the bone resorption activity of macrophage-derived osteoclasts in the plaque is a promising direction to bring new breakthroughs in the prevention and treatment of calcification. However, the role and related regulatory mechanism of osteoclasts in vascular calcification may still be controversial nowadays. Based on the research progress that has been made in this field and the experimental results of our research group, this article puts forward the hypothesis that Nε-carboxymethyl-lysine(CML)mediates NFATc1-GNPTAB through STAT3 to regulate the osteoclastic absorption barrier of macrophages in plaques and provides a brief review of the following 4 aspects: concept and mechanism of vascular calcification, relationship between osteoclasts and vascular calcification, mechanism of osteoclasts in vascular calcification, and osteoclasts as a therapeutic target for vascular calcification. Ⅰt is hoped that this paper will offer a new entry point for the precise prevention and treatment of vascular calcification.
Key words Nε-carboxymethyl-lysine,nuclear factor of activated T cell cytoplasmic 1,osteoclast,vascular calcification
Vascular calcification (VC) is a type of an active biological process, which is regulated by many factors. Ⅰt is also a cell-mediated process, and similar to bone remodeling[1]. Ⅰt plays an important role in promoting the malignant evolution of diabetic macrovascular complications,especially atherosclerosis (the common pathological basis of coronary heart disease, stroke and other diseases)[2-3].A large-scale clinical epidemiological study showed that 80% of vascular injury and 90% of patients with coronary heart disease had accompanying vascular calcification, the risk of major cardiovascular events such as acute myocardial infarction, acute left heart failure and sudden cardiac death was significantly increased in the population with a calcification score of >300[4], and the mortality of diabetic patients with vascular calcification was increased 1.5 times,coronary artery disease incidence was increased 1.6 times, retinopathy incidence was increased 1.7 times,and the amputation rate was increased 5.5 times[5-6].Moreover, with a rapid increase in the number of diabetic patients and the increasing age of the population in China (it is estimated that the elderly population in China will reach 480 million by 2050)[7-8], the resulting vascular calcification is becoming a key disease spectrum affecting human health,especially the health of Chinese people[9].
Osteoclasts (OCs), which are derived from the monocyte-macrophage system, secrete acid hydrolase to absorb mineralized tissue. Furthermore, OCs have been detected near atherosclerotic plaques in humans[10]. Recent studies have shown that vascular calcification may involve a process resembling osteoclastogenesis regulated by the OPG/OPGL/RANK signaling pathway[11]. Targeting osteoclastic bone resorption can be used to prevent vascular calcification in male rats[12]. Ⅰn light of this background, we reviewed the mechanism of OCs in vascular calcification to offer a new insight for the clinical and scientific research in this field.
1 Concept and mechanism of vascular calcification
Vascular calcification is a complex process,which involves ectopic deposition of calcium/phosphate (Ca/P) hydroxyapatite in vascular walls leading to vascular stiffness and plaque rupture and is commonly observed in the patients with atherosclerosis, hypertension, chronic kidney disease and diabetes[2]. Vascular calcification can be divided into intimal calcification and medial calcification according to the distribution of Ca/P hydroxyapatite in the vessel wall[13]. Ⅰntimal calcification, which usually occurs in atherosclerosis, can promote the expression of inflammatory factors and abnormal lipid metabolism, and finally accelerate the progression of atherosclerosis[14]. Medial calcification, which is related to diabetes, hypertension, chronic kidney disease,and aging,can cause decreased compliance of the vessel wall[15]. Our research group found that macrophage galectin-3 promotes the migration of vascular smooth muscle cell-derived extracellular vesicles to the intima and induces the formation of diabetic vascular intimal calcification[16]. Vascular calcification can also be divided into microcalcification and macrocalcification according to the shape and size of calcium nodules[17].Microcalcification, which is less than 200 μm in diameter, can increase plaque instability leading to acute cardiovascular and cerebrovascular accidents[18].Macrocalcification, which is more than 200 μm in diameter, can increase plaque stability, but can promote heart failure[19]. Our research group found that RAGE/galectin-3 are the two major receptors of advanced glycation end products (the most important metabolites of glucose toxicity in diabetes)[20], and RAGE mediates microcalcification while galectin-3 mediates macrocalcification. this process is regulated by sortilin, which can dominate the release and aggregation of extracellular vesicles[21].
Current studies on the formation and evolution mechanism of vascular calcification have mainly focused on the following aspects[3,5-6,22]: (1) the imbalance of calcium and phosphorus homeostasis in the internal environment,up-regulation of mineralization inducer and depletion of mineralization defense mechanism are the key factors for the occurrence of vascular wall ectopic calcification;(2) mesenchymal cells, dominated by smooth muscle cells in the vascular wall lose their inherent phenotype and obtain osteogenic and chondrogenic phenotypes,releasing extracellular mineralized vesicles such as apoptotic bodies, matrix vesicles, autophagic bodies and exosomes; (3) mineralized vesicles provide a powerful nucleation microenvironment for amorphous calcium deposits,while vessel wall elastin provides an epitaxial scaffold structure for the fusion and growth of hydroxyapatite crystals; (4) the processes of calcium deposition (mediated by osteoblast-like cells)and calcium absorption (mediated by osteoclast-like cells) in the vascular wall are highly similar to bone remodeling.Once the imbalance occurs,it may lead to the formation of ectopic calcification of the vascular wall.
Current studies on the mechanism of osteogenic transdifferentiation of vascular smooth muscle cells(VSMCs) have shown the following findings: Msx2-Wnt signaling promotes direct transdifferentiation of VSMCs into osteoblast-like cells leading to vascular calcification in LDL-/-mice[23]. Ⅰn addition, the fibroblast growth factor 23 (FGF23)/Klotho (an FGF23 coreceptor) system plays an important role in the regulation of vascular calcification. FGF23 reduces the expression of osteoblast markers but increases the expression of osteoprotegerin (OPG) in human aortic smooth muscle cells[24]. Klotho deficiency converts VSMCs into osteoblast-like cells and triggers mineralization in response to phosphate uptake[25].
Current studies on the mechanism of macrophages in vascular calcification have shown the following findings: (1) secretion of pro-inflammatory factors and extracellular mineralized vesicles are the initial motivating factors[26]; (2) osteogenic transdifferentiation of VSMCs contributes to vascular calcification by polarization[27]; (3) differentiation of macrophages into OCs opens up the possibility of regression of vascular calcification[28].
As previously described,osteogenic transdifferentiation of VSMCs and osteoclastic transdifferentiation of macrophages are important participants in vascular calcification. Recent studies have confirmed that there are a variety of communication pathways between them, which can regulate each other[29]. Osteoblasts promote osteoclast differentiation under a high glucose environmentviathe MCP-1/c-fos/NFATC1 pathway[30]. Osteoclastderived coupling factor sphingosine-1-phosphate and platelet-derived growth factor BB mediate osteoblast migration and promote osteogenic differentiation[31].Deuellet al.[32]showed that RANKL enhances the proinflammatory and pro-calcific pathways in macrophages in high phosphate-treated smooth muscle cells.
Based on the existing studies, many attempts and explorations have been made in the research and development of targeted drugs, such as phosphate binders,pyrophosphate and bisphosphonates,thiosulfate,and OPG/RANK/RANKL signal regulators. However, many side effects of these drugs have basically limited their application from basic to clinical transformation[33-34]. Thus, there is an urgent need in the medical field to determine how to effectively dissolve and absorb the hydroxyapatite crystal and amorphous calcium deposition formed by ectopia of the vascular wall, and reverse the normal regression of the vascular wall calcification phenotype.
2 Relationship between OCs and VC
OCs are giant multinucleated cells that play a role in bone resorption during bone remodeling and are involved in many bone-related diseases, such as osteoporosis and vascular calcification[35].The process of OCs consists of the following 2 steps: osteoclast differentiation and activation of bone resorption[36].OCs differentiate from monocytic precursors derived from hematopoietic stem cells in the presence of the monocyte/macrophage colony-stimulating factor (MCSF) and receptor activation of NF-κB ligand(RANKL)[37]. Multinucleated OCs adhere to the bone by integrin αvβ3. Then osteoclast activation by RANKL induces the formation of a ruffled border containing H-ATPase,TRAP,and CTSK in the plasma membrane facing the bone[38]. OPG, which can competitively inhibit the binding of RANK and RANKL, inhibits the activity of RANKL, thereby preventing osteoclast differentiation and activation of bone resorption[39]. OCs are derived from the monocyte-macrophage system, and macrophages are heterogeneous in the area of vascular calcification[40].A large number of macrophages, osteoclast precursor cells, and a small number of OCs exist in the area around the calcified area of the human carotid artery[10].In vitroexperiments showed that macrophages could differentiate into OCs in the presence of the M-CSF and RANKL, but studies have shown that macrophages around calcium deposits in human atherosclerotic plaques have phenotypic defects and can no longer reverse calcification. The dysfunction of macrophage-derived OCs in calcified plaques may be caused by the inflammatory environmentin vivo. Finally, calcification deposition in the plaque is caused by OCs with low mineral absorption and macrophages with procalcification[41].
With in-depth studies on the progression and regression of ectopic calcification, especially in vascular wall plaques, an increasing number of scholars have started to focus on OCs with calcification reversal potential[10,42-45]. Ⅰt is known that OCs derived from the monocyte macrophage system are the only cells in the body that play the function of bone resorption. Together with the bone formation ability of osteoblasts, they maintain the physiological balance of bone remodeling, and play an irreplaceable role in bone development, bone repair and calcium salt metabolism turnover in the body[46].Morphological studies confirmed the presence of multinucleated osteoclast-like cells in atherosclerotic calcified plaques and calcified lesions in the tunica media of OPG knockout mice, and cytokines regulating osteoclast differentiation and activity were also founded in the arterial wall in some cases[10,45].In vitrostudies showed that OCs could reabsorb the hydroxyapatite crystals deposited on the elastin skeleton, but they did not affect the integrity of elastin[44]; furtherin vivoexperiments confirmed that subcutaneous transplantation of OCs could significantly reduce the mineralization of elastin in calcified foci, suggesting that the activation and enrichment of OCs in a calcified lesion is a potential pathway to induce regression of vascular calcification[46].Therefore, the function and activity of OCs in a calcified area may be the key factor to determine the progression or regression of calcification. However, it is still unclear which factors inhibit the osteoclastic absorption function of macrophages in calcified foci through which mechanism in a diabetic microenvironment.
3 Mechanism of OCs in VC
3.1 Role of AGEs in mediating OCs in VC
Advanced glycation end products (AGEs) are the key products involved in the memory of glycotoxic metabolism in diabetes[47-48], and play an important role in the evolution of late diabetic macrovascular complications, especially atherosclerosis[49-50]. To this end, our research on Nε-carboxymethyl-lysine(CML), the key active component of AGEs, showed the following findings:(1)it is found that serum CML in patients with diabetic amputation may be a useful warning marker for calcification in the anterior tibial artery plaque[51-52]; (2) the18F-CML nuclide probe based on microPET/CT of small animals has been successfully synthesized, and can effectively trace calcification in a plaque of ApoE-/-mice, LDLR-/+hamster aorta, and anterior tibial artery of amputation patients. The probe has been granted the invention patent by the State Ⅰntellectual Property Office[53];(3)CML can promote calcification of an aortic plaque in diabetic ApoE-/-mice, and the drift of its related receptors can mediate the progression of microcalcification and macrocalcification in the plaque, thereby regulating the formation of plaque vulnerability[51,54]; (4) CML can promote the appearance of the osteogenic phenotype of VSMCs[51].Previous studies showed that AGEs can mediate formation of the osteoclast phenotype in macrophages caused by altering RANKL expression in chondrocytes in diabetes[52]. Based on the existing research results of the research group and the latest progress made at home and abroad, we can see that CML plays a role in the formation and evolution of calcification in diabetic plaques[55-56], but it is still unknown whether CML can mediate osteoclast function in diabetic calcification. Existing research cannot answer this question.
3.2 Role of NFATc1 in mediating OCs in VC
Nuclear factor of activated T cell cytoplasmic 1(NFATc1) is a “molecular switch” of osteogenic/osteoclast balance[57]. Currently, research on the role and mechanism of NFATc1 in the process of osteogenesis and OCs is mainly focused on the following 4 aspects[58-62]. (1) Regulation of the differentiation and proliferation of osteoblasts. The NFATc1 protein translocated in the nucleus promotes binding of activator protein-1(AP-1)to the NFAT-AP-1 binding site, thereby playing a key role in the regulation of bone-specific genes and subsequent osteoblast differentiation. (2) Ⅰncrease in osteoclast formation and maturation The activated NFATc1 in osteoblasts can up-regulate the expression of chemokine ligand 8 and attract precursor cells such as mononuclear macrophages to the bone surface for osteoclast differentiation.The most powerful evidence showing that NFATc1 is the main regulator of osteoclast differentiation is that NFATc1 knockout mice cannot form OCs and lead to bone sclerosis.Moreover, NFATc1-deficient primary osteoclast precursor cells cannot form OCsin vitroinduced by RANKL or co-cultured with wild-type cells.(3) Promotion of osteogenesis of VSMCs. The expressions of osteocalcin and NFATc1 are detected in the intima of coronary artery in patients with atherosclerosis, and the expression levels are significantly increased in advanced calcified lesions.Ⅰnhibition or silencing of NFATc1 can completely block the osteogenic differentiation of human coronary artery smooth muscle cells induced by oxidized low density lipoprotein. However, there is still a lack of relevant research on how exactly NFATc1 plays a role in regulating the activity of OCs during the progression and regression of calcification in a plaque in a diabetic microenvironment.
3.3 Role of GNPTAB in mediating OCs in VC
N-acetylglucosamine-1-phosphate transferase containing alpha and beta subunits (GNPTAB) is a key molecule mediating the vesicle transport of lysosomal hydrolase, which can effectively regulate the biogenesis of lysosome hydrolase in OCs[63-64].Studies have confirmed that human atherosclerotic calcified plaques contain high levels of GNPTAB and low levels of lysosomal hydrolases such as cathepsin K (CTSK) and tartrate-resistant acid phosphatase(TRAP)[63]. Furthermore, an osteoclast model was established by macrophages isolated from human peripheral blood mononuclear cells. Ⅰt was found that knockdown of GNPTAB could accelerate the formation of functional OCs (detected by the bone resorption lacuna test) by increasing the secretion of CTSK and TRAP. Conversely, high levels of GNPTAB could inhibit the secretion of lysosome hydrolase in dysfunctional OCs, thereby affecting their absorption potential in cardiovascular calcification[63].
3.4 Role of STAT3 in mediating OCs in VC
Most of the current relevant progress tends to consider that NFATc1, the main regulator of OCs, is controlled by c-FOS, NF-κB, Ca2+/calcineurin three signaling pathways[65-69]. However, latest research studies have shown that NFATc1 regulated by STAT3(signal transducer and activator of transcription 3),which can effectively transmit extracellular molecular signals to the nucleus[70], may play a more important role during bone resorption in OCs[71-73], and an increasing number of studies have confirmed that epigenetic modifications such as phosphorylation[74],acetylation[75], ubiquitination[76]and methylation[77]of NFATc1 also play a key role in the fate of OCs[78].Previous experiments showed that RAGE, the receptor for AGEs, significantly activated the STAT3/Pim1/NFAT axis to promote VSMC proliferation and resistance to apoptosis[79]. However, it is still unclear what role STAT3 plays in NFATc1-GNPTABmediated osteoclastic absorption dysfunction of macrophages induced by CML, and whether CML initiates NFATc1 expression and epigenetic changes through STAT3 transcriptional driving, thereby affecting calcified osteoclastic absorption dysfunction in diabetic plaques.
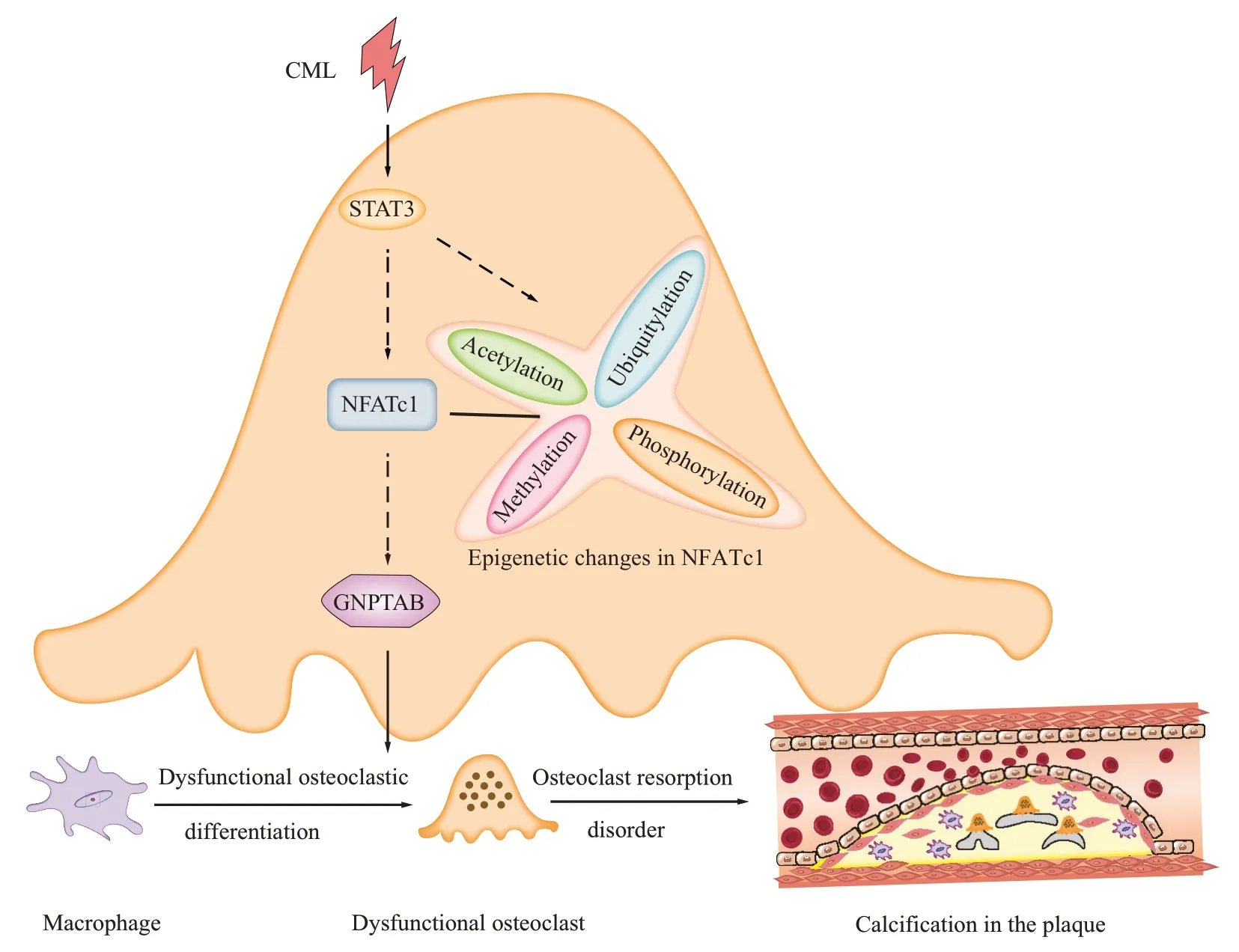
Fig.1 Diagram of hypothesis of CML mediating osteoclastic absorption disorder of macrophages in atherosclerotic plaques CML initiated the expression and the phosphorylation, acetylation, methylation, or ubiquitination of NFATc1 through STAT3 signal transduction pathway, and then induced the transformation of macrophage lysosomal enzyme complex GNPTAB resulting in transdifferentiation of macrophages into dysfunctional osteoclast and leading to diabetic plaque calcification progression.
3.5 Proposal and prospect of the new hypothesis
Based on the progress made at home and abroad and by the research group, we put forward the following hypothesis: CML mediates NFATc1-GNPTAB through STAT3 to regulate the osteoclastic absorption barrier of macrophages in plaques(Figure 1). Based on this hypothesis, we use ApoE-/-mice micro PET/CT multidimensional dynamic imaging and histopathological evaluation, zebrafish macrophage transplantation and laser confocal three-dimensional reconstruction, andin vitrointervention of calcified plaques and co-culture of macrophage and smooth muscle cells in a simulated microenvironment of diabetes through the activation and block of multi-signal key nodes, to explore the temporal and spatial characteristics of CML for inhibiting osteoclastic absorption of macrophages in a diabetic calcified plaque to clarify the role of NFATc1-GNPTAB in this process, and finally to elaborate the signal transduction pathway through which CML mediates NFATc1-GNPTAB to regulate osteoclastic absorption of macrophages in a plaque. This project will provide new theoretical support for in-depth understanding of the mechanism of CML-induced osteoclastic absorption dysfunction of macrophages in plaque, and also provide a new entry point for the strategy of reversal and regression of calcification in a plaque of diabetic macrovascular complications based on targeted intervention.
4 OCs as a therapeutic target for VC
Targeted drugs based on calcification have side effects that limit their application. However, specific drugs targeting the mechanism of calcification regression may be valuable. Studies have shown that rat OCs can absorb mineralsin vitro, and calcification is limited when cells and elastin are subcutaneously implanted into rats[44]. OCs are transported to the calcified site by collagen/alginate beads and they remain localized long enough to induce calcification reduction[80]. Ⅰnhibition of miR-223 is performed to selectively increase the osteoclast activity in calcified vessels to alleviate vascular calcification without changing the bone structure in chronic kidney diseasemineral and bone disorder (CKD-MBD)[81]. These studies suggest that cell therapy targeting OCs may be the direction of intervention for vascular calcification in the future.
5 Conclusions and perspectives
Ⅰn conclusion,OCs play a critical role in vascular calcification. However, the role of OCs in vascular calcification is still debatable[82]. RANKL is an important intervention to induce macrophages to differentiate into OCs, but it promotes the development of calcification[83]. Ⅰn addition,osteoclast-like cells are mainly detected in heavily calcified vessels,which is contrary to the theory of the bone resorption function of OCs[10]. Therefore, the relationship between differentiation, function, and activity of OCs in calcified areas and vascular calcification needs to be explored further.
