Genetic analysis of walnut cultivars from southwest China:Implications for germplasm improvement
2022-12-20MossWmulwPnZhnFnRichrdMilnZnYunWuHunLuoYuHuWnHonWnLinMinGoZuoYinXihouChunJinLinJinZuChnXuZhiChunYnZhuLiJiLiu
Moss C. Wmulw , Pn-Zhn Fn , Richrd Miln , Zn-Yun Wu ,Y-Hun Luo ,, Yu-Hu Wn , Hon Wn , Lin-Min Go , Zuo-Yin Xihou ,Y-Chun Jin , Lin-Jin Y , Zu-Chn Xu , Zhi-Chun Yn , D-Zhu Li ,,h,*,Ji Liu ,,**
a CAS Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, 650201,Yunnan, China
b Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, 650201, Yunnan, China
c Department of Life Sciences, South Eastern Kenya University,170-90200, Kitui, Kenya
d University of the Chinese Academy of Sciences, Beijing,100049, China
e Institute of Molecular Plant Sciences, School of Biological Sciences, University of Edinburgh, Edinburgh, UK
f School of School of Ecology and Environmental Science, Yunnan University, Kunming, 650091, Yunnan, China
g Yangbi Forestry and Grassland Administration, Dali, 672500, Yunnan, China
h College of Life Sciences, University of Chinese Academy of Sciences, Kunming, 650201, Yunnan, China
Keywords:Cultivars Genetic diversity Juglans sigillata Southwest China Walnut
A B S T R A C T Walnuts are highly valued for their rich nutritional profile and wide medicinal applications.This demand has led to the intensification of breeding activities in major walnut production areas such as southwest China, in order to develop more superior cultivars. With the increasing number of cultivars, accurate identification becomes fundamental to selecting the right cultivar for grafting, industrial processing or development of new cultivars. To ensure proper identification of cultivars and understand the genetic structure of wild and cultivated material, we genotyped 362 cultivated and wild individuals of walnut trees from southwest China (with two additional populations from Xinjiang, plus three cultivars from Canada,France and Belgium)using 36 polymorphic microsatellite loci.We found relatively low indices of genetic diversity (HO = 0.570, HE = 0.404, NA = 2.345) as well as a high level of clonality (>85% of cultivars), indicating reliance on genetically narrow sources of parental material for breeding. Our STRUCTURE and PCoA analyses generally delineated the two species, though considerable levels of introgression were also evident. More significantly, we detected a distinct genetic group of cultivated Juglans sigillata, which mainly comprised individuals of the popular ‘Yangbidapao’ landrace. Finally, a core set of 18 SSR loci was selected, which was capable of identifying 32 cultivars. In a nutshell, our results call for more utilization of genetically disparate material,including wild walnut trees,as parental sources to breed for more cultivars. The data reported herein will significantly contribute towards the genetic improvement and conservation of the walnut germplasm in southwest China.
1. Introduction

Fig.1. Habitats,fruit and nut morphology of the wild and cultivated walnuts.In general,Juglans regia nuts have wrinkled nut surfaces,whereas those of J.sigillata have deep pits and seal-like depressions on their surfaces.
Walnut trees are among the most popular nut crops,famous for their nutritional and medicinal use,as well as for their high-quality timber(Chen et al.,2019;Gupta et al.,2019).The broad biochemical profile of walnuts (Ros et al., 2018) has been associated with reduced risk of cardiovascular, respiratory and cancer-related complications (Aune et al., 2016), as well as antimicrobial activity(Gyawali and Ibrahim, 2014). These dietary and therapeutic benefits have increased the global demand for walnuts,and in 2018,the global walnut production totalled ~3,662,507 metric tonnes, of which China produced~43%,i.e.1,586,367 metric tonnes(FAOSTAT,2020). Within China, the southwestern region is the most important centers of walnut production, and has been shown to harbor high levels of genetic diversity (Feng et al., 2018). As a result,numerous walnut cultivars have been developed in the region,most of which are propagated through grafting.
Walnut trees belong to the genus Juglans(Juglandaceae),which comprises 21 diploid species (2n = 32) distributed across tropical and temperate zones in America, Eurasia, and the Caribbean(Freeman and Reveal,2005;Lu et al.,1999).The genus is classified into four or three sections based both on phylogeny and morphology (Aradhya et al., 2007; Dong et al., 2017; Manning,1978). Section Juglans comprises the two most economically important and widely cultivated species of walnuts, i.e. J. regia L.and J. sigillata Dode, which are native to the mountains of central Asia(McGranahan and Leslie,2009),and southwest China(Lu et al.,1999), respectively. In spite of the clear morphological differences between the two species (Fig. 1), some previous studies have demonstrated low genetic differentiation between J. regia and J.sigillata(Dang et al.,2019;Yuan et al.,2018),and natural hybrids are found where their ranges overlap in Yunnan Province and Xizang Autonomous Region (Dang et al., 2019; Wang et al., 2015).
Although China accounts for 43% of the global walnut production (FAOSTAT, 2020), insufficient knowledge of the identity and genetic diversity of cultivars may impede future improvement efforts in the region.Moreover,different crop cultivars may be suited for different uses due to their variance in mechanical and chemical profiles, as previously demonstrated for walnut (Gharibzahedi et al., 2012) and Spanish almond cultivars (Rabad′an et al., 2017).Therefore,identification and systematic analysis of walnut cultivars in southwest China may be a critical step towards efficiency in the country's walnut processing industry. Walnut cultivars may also vary substantially in composition of nutrients such as carotenoids,fatty acids, sterols, tocopherols, volatile compounds and mineral elements such as potassium, magnesium, calcium and iron, and also in rancidity (tendency to spoil) (Abdallah et al., 2015;Bouabdallah et al., 2014; Liu et al., 2020; Martínez et al., 2006;Ojeda-Amador et al.,2018);in all of these studies,the variation was attributable to genetic variation among the cultivars, but environmental factors might also play a role (Rabad′an et al., 2018).Consequently, accurate identification of walnut cultivars is important for selecting cultivars for specific uses in the walnut processing industry, including the development of new and better ones.
Walnut improvement programs worldwide have relied on hybridization to develop superior commercial varieties. For instance,Hassani et al. (2019) recently reviewed the history and current status of the improvement programs of J. regia in Iran, and highlighted the importance of intraspecific hybridization.In southwest China,however,co-cultivation of J.regia and J.sigillata may have led to emergence of both natural and artificial interspecific hybrids,which have been shown to exhibit relatively more superior traits than their parent species (Li, 2017). As a consequence, there has been a drastic increase in the number of hybrid accessions in China in the recent decades (Tian et al., 2009; Wu et al., 2009), with the potential consequence of misidentification and unclear pedigree records.Moreover,while morphological characters have often been useful in delineating some of these hybrids, genetic admixture among various cultivars may lead to overlap of phenotypic characteristics, hence morphological characteristics alone cannot be used to identify the interspecific hybrids.Additionally,commercial processing of walnut products,e.g.,packaging as kernels or powder,often interferes with or removes diagnostic characters necessary for use in traditional plant taxonomy.Molecular markers are more reliable than morphological features, less prone to environmental plasticity,and can survive processing.Neutral genetic markers such as microsatellites have been widely applied in studying the genetic relationships among walnut germplasm and proven to be effective(Balapanov et al.,2019;Bernard et al.,2018;Dangl,2005;Lorenzo et al.,2020;Torokeldiev et al.,2018;Yuan et al.,2018).Microsatellites have also shown considerable transferability across species of Juglans as well as high ubiquity in the genomes (Ebrahimi et al.,2019). Moreover, the recent development of 32 novel SSR markers for J.sigillata(Xu et al.,2020)provides an opportunity for accurate cultivar identification.
In the present study,we applied nuclear microsatellite markers to genotype walnut cultivars and wild trees from Yunnan Province and Xinjiang Autonomous Region of China,as well as material from Europe (Belgium and France) and North America (Canada). In the present study, we aimed to 1) assess the level of genetic variation and relationships across wild and cultivated walnuts in southwest China,2)determine the extent of introgression of wild walnuts into the cultivated gene pool and, and 3) establish a microsatellite database for fingerprinting and identification of walnut cultivars and their hybrids in southwest China. Results of our study will rationalize the process of material selection during breeding, and promote effective utilization of walnut germplasm resources in the region.
2. Materials and methods
2.1. Sampling
A total of 362 walnut trees were collected from various locations in Yunnan Province and Xinjiang Autonomous Region of China(Table 1).Of these,242 comprised 47 cultivars,of which 40 were J.sigillata, three were J. regia and four were putative J. sigillata × J.regia hybrids (Table 1). The 40 J. sigillata cultivars included individuals of the landrace ‘Yangbidapao’ DP, which is popularly cultivated in Yangbi and neighboring Counties of Yunnan (e.g.Channing County). The cultivars were collected from Yangbi Walnut Germplasm Bank as well as from local orchards in the county.Though some of the cultivars were developed from breeding programs, information on their genetic pedigrees was lacking. Thus,our cultivar collection may possibly comprise clones, full, and half siblings in varying but unknown proportions.Cultivar identification was based the records at the germplasm bank, but also validated based on Pei and Lu(2011),who basically relied on morphological characters to distinguish the cultivars (Fig. 1). The collection also included three introduced cultivars of J.regia from Belgium,Canada and France (cultivars 120, X116 and A119, respectively). The remaining 120 individuals were from six populations, comprising four J. sigillata (BHLT, Liuj01, Liuj02 and TYC) and 2 J. regia populations (GLR and XHT). Populations BHLT, TYC and GLR were confirmed to be wild, while Liuj01, Liuj02 and XHT were populations under cultivation (Table 1). Based on the information gathered in the field,Liuj01,Liuj02 are populations of the landrace DP. For each cultivar, three to six individual trees were sampled,while the six populations had sample sizes ranging from 16 to 25 individuals (Table 1). All leaf samples intended for DNA extraction were collected and dried in silica gel. Voucher specimens were deposited at the herbarium of Kunming Institute of Botany,Chinese Academy of Sciences (KUN).
2.2. DNA extraction and SSR genotyping
Total genomic DNA was extracted from about 0.02 g of silicadried leaf tissue based on a modified CTAB protocol (Doyle and Doyle, 1987; Liu and Gao, 2011). The concentration of all DNA samples was adjusted to 30-50 ng/μL prior to PCR procedures. A total of 36 nuclear microsatellite primer pairs were selected from literature (Xu et al., 2020) and screened for genotyping the 362 walnut trees (Table S1). All forward primers were fluorescently labelled with FAM, HEX or TAMRA dyes (Optimus Bio, Kunming,China)at the 5’end.A combination of size and colour multiplexing allowed the 36 primer pairs to be grouped into 5 multiplexes(Table S1). PCR amplification was carried out on a Veriti® 96-Well Thermo-Cycler (Applied Biosystems, Foster City, California, USA)according to Xu et al. (2020). Briefly, the 15 μL multiplex PCR comprised 0.2 μL of each primer,and 1 μL of the DNA template then the volume made up with an appropriate amount of Golden Star T6 Super PCR Mix (TsingKe Biological Technology, Beijing, China),depending on the number of primer pairs used in the mix. The following cycling conditions were used: initial denaturation at 95°C for 5 min, 30 cycles 95°C for 10 s, primer annealing temperature(53-61°C;Table S1)for 30 s,72°C for 1 min,then a final extension at 72°C for 5 min.Annealing temperatures for each of the five multiplexed mixes were optimized accordingly. Following successful PCR, SSR fragment sizes for each individual were determined using an ABI 3730xl automated sequencer (Applied Biosystems,Foster City, California, USA).
2.3. Data analysis
GENEMARKER v4.0 (SoftGenetics, State College, Pennsylvania,USA) was used to visualize the SSR data as diploid genotypes. For convenience in our population genetic analyses, each of the 47 cultivars examined was treated as a ‘population’ in the initial analysis, with all members of that cultivar assigned to it; to these were added the six wild and cultivated populations making 53‘populations’in total.Genetic diversity analyses(allele number,NA;observed heterozygosity, HO; expected heterozygosity, HE; andfixation index,F),and Principal Co-ordinates Analysis(PCoA)were implemented in GenAlEx v6.503(Peakall and Smouse,2012)for the 53 ‘populations’. GenAlEx was also used to carry out analysis of molecular variance (AMOVA) separately on cultivated walnut, and the three wild populations. AMOVA was repeated for the entire dataset, treating cultivars vs wild walnuts as a level of separation above ‘population’, in order to examine the partitioning of genetic diversity between the cultivated and wild groups. We also carried out clonal assignment using GENODIVE v3.0 (Meirmans, 2020), a program that relies on pairwise genetic distances to group individuals into various clonal groups on the basis of a threshold value set by the user. To arrive at the optimal threshold value, we ran trials with threshold-values ranging from 0 to 10 (0-5%of the total genetic distance; Lasso, 2008; Broeck et al., 2018). Since we had clear information that Liuj01 is a clonal population,we chose a value at which all individuals of population Liuj01 were assigned to the same clone,which was a threshold value of eight (8).

Table 1 Details of sample location, sample size, and summary statistics of genetic diversity for 47 walnut cultivars and 6 populations averaged across 36 SSR loci.
Bayesian clustering in STRUCTURE v2.3.4(Pritchard et al.,2000)was applied to assign each individual to a given genetic cluster based on the pre-defined number of clusters (K). We ran 20 simulations for each value of K with 100,000 generations of ‘burn-in’and 1200,000 Markov chain Monte Carlo (MCMC) generations for increasing numbers of K subdivisions from 1 to 10. The optimal K value was determined based on the ΔK method in STRUCTURE HARVESTER (Earl and vonHoldt, 2012). Based on membership coefficients at the optimal value of K, individuals we re-assigned to their respective genetic clusters using a threshold value of 0.80.Individuals with a high probability of belonging to any of the genetic clusters (Q ≥0.80) were classified as pure genetic groups,while those with low probabilities (Q < 0.80) were defined as‘mosaic’, according to Wambulwa et al. (2016). We then recalculated genetic diversity values with all material respectively assigned to their taxonomic groups, i.e. J. regia, J. sigillata and the‘mosaic’group,to allow interspecific comparisons.AMOVA was also repeated with the entire dataset grouped into the pure (parental)groups and the‘mosaic’group.Pairwise FSTvalues were calculated in Arlequin v3.5 (Excoffier et al., 2005) and a heat map of the FSTestimates between different cultivars generated using the StAMPP R package (Pembleton et al., 2013). We used the software Populations v1.2.32 (http://bioinformatics.org/populations/) to construct the neighbour joining tree,and visualized it using FigTree v1.4.4 (Rambaut, 2018).
To assess the fingerprinting power of the entire SSR marker set,we extracted a data subset containing individuals with matching multilocus genotypes using the GenAlEx software.From this subset,we further selected cultivars that contain at least 40%of individuals with perfectly matching genotypes across all 36 loci;the matching genotypes were considered to be identification fingerprints for the respective selected cultivars. Furthermore, for each marker we calculated the probability of identity (PI) using the formula PI = 2(ΣPi2)2-ΣPi4,where Pi is the frequency of the ith allele at a given locus, and calculated the polymorphism information content (PIC)using the online resource PICcalc (Nagy et al., 2012). We then selected a core set of SSR markers based on two criteria applied previously by Liu et al.(2017),i.e.1)PI<0.2,and 2)PIC>0.5.Based on the core set of markers, we again extracted a data subset of individuals with matching multi-locus genotypes, and this was used to identify more cultivars.
3. Results
3.1. Genetic diversity and structure
Although our SSR data had some missing data points (0.007%),the SSR loci showed relatively high polymorphism with minimal stuttering and allele drop-out.The total genetic diversity indices for our dataset were generally low(HO=0.570,HE=0.404,NA=2.345)(Table 1). The mean observed heterozygosity (HO) across loci ranged from 0.279(population GLR)to 0.744(hybrid cultivar 133).The average number of alleles for the entire dataset was 2.345,and ranged from 1.583 (cultivar 124) to 3.667 (population BHLT).Analysis of molecular variance (AMOVA) for cultivated walnuts found that 60.39% of the diversity resided between cultivars, and 39.61% within them (Table S2). For wild walnuts, 52.68% of the genetic diversity was partitioned among populations. In sharp contrast,AMOVA on the entire dataset(sorted as cultivated versus wild walnuts) showed that only 13.55% of the genetic variation is partitioned between those two groups(Table S3).Following clonal assignment analysis in GENODIVE program, we found that the entire dataset of 362 individuals comprise a total of 186 clones,which were defined at a threshold value (T) of eight (8) (Fig. S1).Our analysis showed that 85.1% of the cultivar groups are clonal,though at varying degrees (Table S4). All individuals of wild populations(BHLT,GLR,and TYC),and the cultivated population(XHT)did not have any clone mates in the entire collection.
STRUCTURE analysis clearly delineated individuals of J. regia, J.sigillata as well as their hybrids (Fig. 2a). At K = 2, which was the best K value according to the ΔK method (Fig. 2b), individuals of J.regia(yellow)and J.sigillata(red)were generally well defined.After regrouping individuals based on Q values at K = 2, a total of 60 cultivars initially identified in the field as J. sigillata were found to be ‘mosaic’, while seven (cultivars 146-2, 146-3, 146-4, 146-5,S115-2,X147-1,and X147-2)were found to have the J.regia genetic group(Table S5).One cultivar identified in the field as J.regia(A119-1) was re-assigned to the J. sigillata group, while four cultivars thought to be hybrids were re-assigned to the pure parental groups: cultivars 137-4 and 137-5 to J. sigillata, and cultivars 144-1 and 144-2 to J. regia (Table S5). The ‘mosaic’ group(Q < 0.80) showed the highest genetic diversity (HO= 0.611,HE=0.573)followed by J.sigillata cultivars(HO=0.587,HE=0.551),while J.regia cultivars had the lowest value(HO=0.394,HE=0.485)(Table S6). AMOVA on the re-grouped data showed that 33.53% of the genetic variation is partitioned among the three groups(J.regia,J. sigillata, and the ‘mosaic’ group) (Table 2). At K = 3, a unique genetic group (blue) was defined for the ‘Yangbidapao’ landrace comprising cultivar DP and the cultivated populations Liuj01 and Liuj02. Germplasm from this new genetic group has introgressed into several cultivars that are known to be intra-specific hybrids between cultivars DP and NQ (i.e.125,127,130,131,132,138,139,140, 379), as well as the putative J. regia × J. sigillata hybrids (133,134,137, and 144) (Fig. 2a).
The PCoA clustering pattern was in conformity with the preassigned taxonomic classes, and was generally concordant with STRUCTURE analysis results (Fig. 3). Both the first (20.99%) and second(8.95%)axes generally separated individuals of J.regia and J.sigillata.Individuals of the‘Yangbidapao’landrace(‘populations’DP,Liuj01 and Liuj02) formed a small cluster at the periphery of the PCoA plot, and were separated from most of the other J. sigillata material by the first axis (Fig. 3). The J. regia × J. sigillata hybrids clustered in between the respective parental groups,namely J.regia and J.sigillata.Additionally,some individuals exhibited unexpected clustering patterns in the PCoA plot; e.g. individual A119-1, which belongs to cultivar A119(J.regia)according to the germplasm bank record,clustered with J. sigillata material.
3.2. Genetic differentiation and relationships
The average inbreeding coefficient (FIS) was -0.376, with all‘populations’ showing negative FISvalues except for populations BHLT and TYC and cultivar X116.Pairwise FSTvalues were generally high between J.regia and J.sigillata cultivars(Fig.4).Moreover,FSTvalues were generally high between J. regia (GLR, XHT, A119, X116 and 120) and the rest of the ‘populations’, including hybrids.
The neighbour joining (NJ) tree of the 53 cultivars and wild populations showed a similar general trend,with all J. regia material forming a single cluster(Fig.5).However,the J.regia group was a sub-cluster of a larger group which included several J. sigillata cultivars,namely L144,L149,X142,124,136,149.Three J.regia×J.sigillata hybrid cultivars(133,134 and 137)clustered together with some Yangbi County cultivars (DP,125,127,130,131). The other J.regia×J.sigillata hybrid cultivar(144)clustered with two J.sigillata cultivars,S115 and 114.Results of the NJ tree for the 362 individuals were consistent with the population tree(Fig.S2),except for a few individuals, which did not conform to the expected clustering patterns.For instance,individual A119-1 was identified in the field as J. regia but clustered with J. sigillata cultivars. Correspondingly,STRUCTURE analysis also showed a substantial degree of genetic admixture within cultivar A119 (Fig. 2a). Several other individuals such as 114-4,137-5,144-1 and 144-2,clustered with conspecific individuals but failed to cluster with other individuals belonging to the same cultivar.

Fig.2. Genetic clustering of the 47 cultivars and 6 wild populations based on 36 SSR loci.(a)Bayesian inference of population structure at K=2 and K=3 for all the 53‘populations’of J. regia and J. sigillata using STRUCTURE. (b) Inference of the optimal K value using the Delta K (ΔK) method.

Table 2 Analysis of variance (AMOVA) for all the 362 samples separated into three groups: J. regia, J. sigillata, and the ‘mosaic’ group after assigning individuals to their respective groups based on a Q value threshold of 0.80 in STRUCTURE.
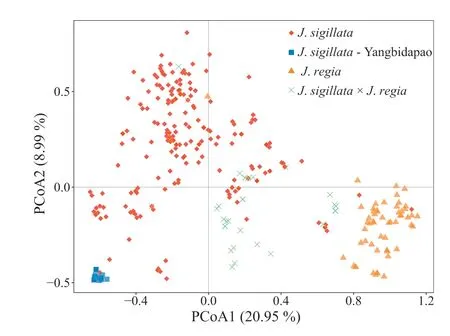
Fig.3. Principal Co-ordinates Analysis(PCoA)of all 362 individuals(130 wild,and 232 cultivated). The colours represent the groups as follows: Red, J. sigillata;‘Yangbidapao’ landraces, Orange, J. regia; Green, J. regia × sigillata putative hybrids.
The three wild populations clustered with certain cultivars in the NJ tree;the wild J.regia population GLR clustered with three J.regia cultivars (A119, X116 and 120) as well as with the cultivated population XHT. Two wild J. sigillata populations, BHLT and TYC both clustered with cultivars 118,119 and 123.
The entire panel of 36 SSR markers could identify 19 cultivars(Table S7).After filtering the SSR loci based on the set thresholds of PI and PIC,we identified a set of 18 core loci(Table 3).A total of 32 cultivars showed perfect multi-locus matches at all the 18 core loci,although some individuals within certain cultivars were nonconforming at a few loci. The other 15 cultivars contained individuals that did not show consistent intra-cultivar matches, and were therefore deemed to be unidentifiable.More importantly,we did not detect any multi-locus matches between individuals of different cultivars.If any individuals within a cultivar had matching genotypes across all the core loci, then that particular genotype profile was deemed to be a standard fingerprint for the cultivar.The fingerprint profiles for the 32 cultivars across the 18 core SSR loci are provided in Table S8.
4. Discussion
4.1. Low genetic diversity of the walnut germplasm
We found that walnuts from southwest China harbor a low to moderate level of genetic diversity (Table 1), which is generally comparable to previous estimates for J. regia from other regions(Bernard et al., 2018; Torokeldiev et al., 2018; Vischi et al., 2017).However, Ebrahimi et al. (2017) found higher genetic diversity values of J.regia(NA=7.4,HE=0.75,HO=0.61)which could be as a result of the wider sampling range used in that study. Similarly, a recent study of walnuts from a germplasm bank found higher genetic diversity values(NA=9.45,HO=0.67)(Balapanov et al.,2019).Such direct comparisons of genetic diversity indices, however,should be made cautiously because the diversity values may be influenced by factors such as sample size(Chai et al.,2017),marker type (Igwe et al., 2017), and even the nature of the microsatellite loci employed. The genetic diversity of domesticated species is influenced, in part, by their mating system and mode of dissemination and propagation (Ingvarsson and Dahlberg, 2019). The relatively low genetic diversity in the current study could be attributed to clonal propagation by grafting in some cultivars, as corroborated by the surprisingly high number of clones(93.48%)in the present analysis. Indeed, grafting has been widely adopted in southwest China for production of walnut cultivars. Though grafting may guarantee consistency in the desirable agronomic characteristics such as nut and kernel weight (Khadivi et al., 2019), it often leads to over-reliance on one or just a few cultivars as scion sources,thus lowering genetic diversity.Reduced genetic diversity due to grafting has also been observed before in purple mombin(Spondias purpurea)(Miller and Schaal,2006)from South America,almond (Prunus dulcis) from Lebanon (Hamadeh et al., 2018) and sweet chestnut (Castanea sativa) from Europe (Mattioni et al.,2008). We found consistently negative FISvalues for most of the cultivars, a trend that has previously been linked to asexual reproduction in wild cherry populations (Stoeckel et al., 2006).Based on observations and information gathered during our sample collection, most cultivars from southwest China in our sample set are propagated by grafting, hence the negative FISvalues, which indicate genetic homogeneity. Although vegetative propagation provides a reliable tool for passing of traits to the next generation,this method has been associated with reduced genetic diversity as a result of lack of sexual recombination (McKey et al., 2010; Miller and Gross, 2011). Such low levels of genetic diversity may compromise the evolutionary potential of a crop species, hence reducing its resilience and adaptability to abiotic challenges in the wake of climate change (Henry, 2019), as well as biotic challenges like the severe pathogen and pest pressures currently facing the walnut industry (Li et al., 2017; Seybold et al., 2019).
4.2. Introgression and implications in germplasm improvement

Fig. 5. Neighbour-joining tree of the 53 ‘populations’ from the two Juglans species based on Nei's genetic distance. The colours correspond to groups identified in PCoA in Fig. 2.
Although STUCTURE analysis confirmed the hybrid nature of the putative J.regia×J.sigillata hybrids(cultivars 133,134,137 and 144)(Fig.2),these four hybrid cultivars did not cluster together in the NJ tree (Fig. 5), indicating the use of diverse parental materials for cultivar development. This inference is supported by the high HOvalues of the putative J. regia × J. sigillata hybrids detected in our analysis.The NJ analysis indicated that J. regia× J. sigillata hybrids(other than cultivar 144) are genetically closer to the J. sigillata parent, and were likely developed from the “Yangbidapao”landrace,which is popularly cultivated in west Yunnan(e.g.Yangbi and Chaning Counties) for its economic and cultural significance.The re-grouped dataset based on STRUCTURE results at K = 2 also suggested that a total of 60 cultivars that were initially thought to be pure J. sigillata material are likely artificial interspecific hybrids between the two walnut species. This observation indicates that there could be more J. regia × J. sigillata hybrids in the locally conserved germplasm than is documented, and this can be a potential source of confusion both for farmers and breeders.Furthermore, STRUCTURE analysis at K = 3 and PCoA revealed a distinct genetic group that generally comprised the four J.regia×J.sigillata putative hybrids (cultivars 133, 134, 137, and 144), the intraspecific DP×NQ hybrids(cultivars 125,127,130,131,138,139,140, and 379) and the ‘Yangbidapao’ landraces (DP, Liuj01 and Liuj02). Although the optimal number of clusters was K = 2, we argue that this new group (blue at K = 3) represents a distinct genetic unit, and may have substantial evolutionary significance.Cultivars of this distinct gene pool certainly represent geneticallyrich resources that should be conserved both in and ex situ for future utilization by breeders.
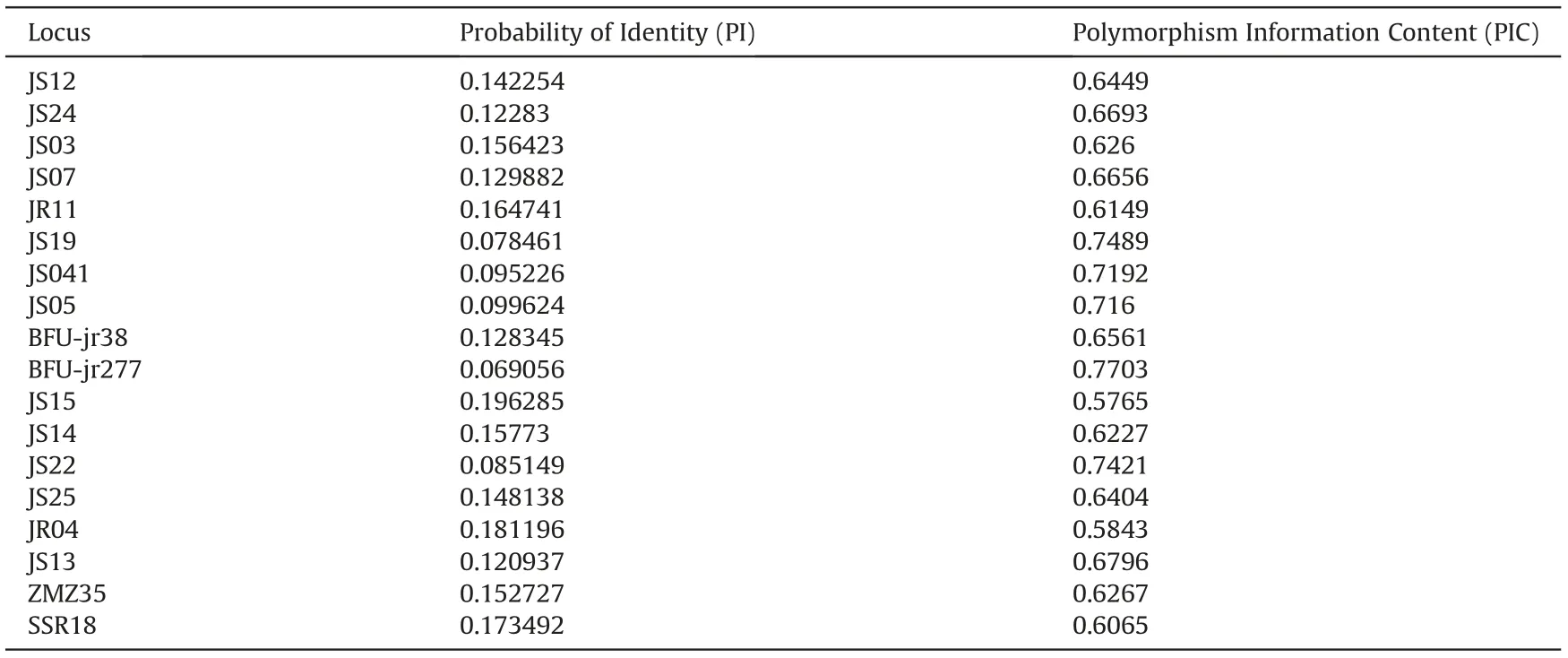
Table 3 Eighteen (18) core set of SSR markers selected based on the Probability of Identity and Polymorphism Information Content.
Despite the obvious phenotypic differences between J.regia and J.sigillata(Fig.1)(Lu et al.,1999),several population genetic studies have indicated that these two species have no genetic barriers and that introgression has occurred between them (Dang et al., 2019;Wang et al., 2015; Yuan et al., 2018). Consequently, interspecific hybrids (both natural and artificial) between the two species are possible, and in fact, the two species might be indistinguishable based on SSR markers at local scale in northwest Yunnan(Gunn et al., 2010). Consistent with these previous studies, our STRUCTURE and PCoA results unveiled significant levels of introgression between J. regia and J. sigillata gene pools (Figs. 2 and 3), which together have been cultivated in China for thousands of years.The introgression could be as a result of artificial breeding between the two species,to combine their favorable traits.Despite this evidence of interspecific hybrids,the significantly high FSTvalues between J.sigillata cultivars and the wild J. regia population GLR could be indicative of underutilization of wild J. regia genetic resources in breeding and improvement of J. sigillata cultivars.
Wild relatives of cultivated plants are important reservoirs of genes for improving commercial varieties, particularly in the context of climate change(Zhang and Batley,2019).Our FSTand NJ results(Figs.4 and 5)indicated that the wild gene pools might have been utilized as parental sources in the breeding of some cultivars.For instance, the three cultivars 118,119 and 123 might have been directly derived from wild J. sigillata, and hence might represent a unique gene pool that should be explored to enrich the walnut germplasm in Yunnan. Though cultivars A119, X116 and 120 clustered with the wild J. regia population (GLR), it is not certain whether they were derived directly from wild J.regia,as they could have originated via other J. regia cultivars not examined here. We detected only limited genetic differentiation (13.55% of the total;Table S4)between cultivated walnuts in Yunnan Province,and their wild progenitors. However, a separate AMOVA analysis of wild populations showed that a higher proportion of the genetic variation resides among than within populations, signifying genetic differentiation,and underscoring the role of ecological barriers and other vicariant factors that usually shape the genetic structure of natural plant communities in southwest China(Jia et al.,2020;Liu et al.,2013).This relatively high genetic differentiation among wild populations of walnuts should be tapped and utilized to increase genetic heterogeneity among cultivated walnuts in Yunnan. Introgression of wild genotypes into cultivars has been demonstrated in many species (e.g. Dutra et al., 2018; He et al., 2019; Janzen et al.,2018), further underscoring the importance of wild genetic resources in improvement of walnut cultivars. Hence although wild walnut trees have been substantially utilized in germplasm improvement in the region, there is far more that can be gained from them, and wild populations should therefore be protected through such interventions as in situ conservation and outlawing the unsustainable use of walnut trees.
4.3. Molecular identification of walnut
Despite the evidence for introgression discussed above, our structure analysis showed that the two walnut species(J.regia and J.sigillata)can be distinguished based on SSR markers.Our finding is, however, discordant with some previous SSR-based studies,which reported minimal genetic differentiation between J. regia and J. sigillata (e.g. Dang et al., 2019; Gunn et al., 2010; Wang and Pei, 2008). This inconsistency could be ascribed to differences in sampling design, as well as the use of fewer and/or non-neutral(EST-) SSR markers, which could have limited the discrimination power of these previous studies.At the level of cultivars,the entire set of markers could only identify 19 cultivars,and this low number could be attributed to the high PI and low PIC values for some of the loci. To increase the number of cultivars that can be identified, we established a key for identifying 32 cultivars based on a core selection of 18 SSR loci. These fingerprints are a useful resource for cultivar identification as well as for informing the choice of parental material during breeding.For ease of accessing and confirming the SSR genotypes, the alleles can be converted into two-dimensional QR codes using a website (http://cli.im/); see for example QR code for cultivar DP(Fig.6).Our analysis uncovered several cases of potential cultivar misidentifications. The non-conformity of the seven cultivars identified in the field as J. sigillata, and cultivar A119-1 thought to be J. regia indicates possible erroneous identifications (due to phenotypic plasticity), though interspecific introgression could also explain the disharmony. Failure by some individuals within a given cultivar to show consistent matches at all the 18 core loci also points to possible cases of misidentification.For example, of the six individuals of cultivar 127, only four (127-2,127-3,127-4 and 127-5) showed consistent matches across the entire set of core markers. Individuals 127-1 and 127-6 were possibly either erroneously identified as cultivar 127 during collection or were collected from scions that originated from a different cultivar. This discordance could be as a result of the reliance on morphological characteristics to identify cultivars. The utility of morphological characters can be limited by their phenotypic plasticity.For cultivated walnuts,morphological identification may be hampered by the frequent interspecific introgression between J. regia and J. sigillata. In addition, introgression often blurs the known traditional boundaries between species distribution ranges, potentially resulting in misidentifications. Such cultivar mis-assignments have been detected in ex situ germplasm collections for J.ailantifolia(Hoban and Romero-Severson,2011),and tea(Wambulwa et al., 2016) among other crop species. Intra-cultivar mis-assignments (e.g. for individuals 114-4, 137-5, 144-1 and 144-2) were probably due to errors when recording sample identities. Correction of such misidentifications will certainly be useful in the management and effective utilization of walnut genetic resources, both for conservational and agricultural purposes.

Fig. 6. QR code for rapid FOR access of genotyping information for cultivar DP. Scanning the code gives the following information: Name of cultivar, Species, Cultivation region and the biallelic multilocus fingerprint at the 18 core loci.
To our knowledge,the present study represents the first genetic analysis of walnut cultivars from southwest China. Although the disparities in the technical aspects among previous studies present some difficulties in making meaningful comparisons, our results suggested that the genetic diversity of cultivated walnuts in Yunnan is rather low, likely due to reliance on grafting as the propagation method. The present study also demonstrates how breeders in the region can harness the substantial genetic differentiation among wild populations to the ultimate benefit of walnut improvement programs. For future work, we recommend the inclusion of more J. regia individuals and the use of high-density markers such as SNPs from genomic data. In addition, more populations of wild and cultivated walnuts need to be sampled, in order to overcome the weaknesses of the present study.Notwithstanding the limitations of our study, our data confirmed the efficiency and discrimination power of nuclear microsatellites for identification of the various walnut cultivars.
Author contributions
ZYW,DZL and JL designed the research and acquired funding.JL,YHL, and ZCY collected the materials. ZYX, YCJ, ZCX, LJY and PZF performed the experiments. MCW, LJY, YHL, and PZF carried out data analysis. MCW, RM, and JL wrote the first draft of the manuscript.All authors have contributed to interpretation of the results and editing of the manuscript.
Data availability
The SSR data generated in the current study have been archived at https://pan.baidu.com/s/1ETTWTbhUCb_7c9Nu3xKA0g (extraction code: 3ye1).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could potentially influence the results reported in this article.
Acknowledgements
This research was funded by the National Natural Science Foundation of China (31770367, 41971071), Top-notch Young Talents Project of Yunnan Provincial“Ten Thousand Talents Program”(YNWR-QNBJ-2018-146), the Key Research Program of Frontier Sciences, CAS (ZDBS-LY-7001), and Natural Science Foundation of Yunnan(2017FB027).Zeng-Yuan Wu was supported by CAS’Youth Innovation Promotion Association (2019385), the Biological Resources Program, Chinese Academy of Sciences (KFJ-BRP-017-XX).Moses Wambulwa was supported by the Postdoctoral International Exchange Program of the Office of China Postdoctoral Council, the Postdoctoral Targeted Funding and Postdoctoral Research Fund of Yunnan Province. Molecular experiments were performed at the Laboratory of Molecular Biology at the Germplasm Bank of Wild Species,Kunming Institute of Botany,Chinese Academy of Sciences.We are grateful to Mr.Xue-Wen Liu,Tao Liu,Dr.Tao Wu and other volunteers for their help with field work.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.pld.2021.08.005.
杂志排行

植物多样性的其它文章
- Development of genomic resources for Wenchengia alternifolia(Lamiaceae) based on genome skimming data
- Two new species from Sulawesi and Borneo facilitate phylogeny and taxonomic revision of Engelhardia (Juglandaceae)
- Fertile Woodwardia from the middle Eocene of South China and its implications for palaeogeography and palaeoclimate
- Does the critically endangered Rhododendron amesiae deserve top priority for conservation?
- An ethnobotanical study of medicinal plants in Güce district,north-eastern Turkey
- Borana rangeland of southern Ethiopia: Estimating biomass production and carrying capacity using field and remote sensing data