溶酶体代谢酶甘露糖-6-磷酸糖基化修饰关键酶UCE 的异源表达及功能鉴定
2022-12-09曹筠嵩杨燕刘忞之王伟
曹筠嵩,杨燕,刘忞之,王伟
溶酶体是具有单层膜的细胞器,其内部呈酸性,含有 60 多种酸性的水解酶,用于降解生物体代谢过程中产生的各种生物大分子,包括蛋白质、核酸、脂质、多糖等,并且参与细胞自噬的过程。溶酶体代谢酶在翻译后需要在 N-糖基位点(N-X-S/T,X ≠ P),即特定的天冬酰胺残基被糖基化修饰形成甘露糖-6-磷酸(mannose 6-phosphate,M6P)的寡糖结构,才能够被转运到溶酶体发挥功能[1]。用于治疗溶酶体贮积症的替代酶类药物,大多由哺乳动物细胞表达,其 M6P 含量往往不足,影响其靶向溶酶体的转运,导致治疗效率降低。
溶酶体代谢酶的生物合成是在内质网向高尔基体转运过程中首先形成 Man8GlcNAc2(即 8 个甘露糖,2 个 N-乙酰葡萄糖胺)的寡糖结构,然后被转运到高尔基体中继续修饰[2-3]。在高尔基体中形成 M6P 结构依赖于两个关键酶的作用,第一个糖基化修饰反应是由定位于顺面高尔基体(cis-Golgi)的 N-乙酰葡萄糖胺-1-磷酸转移酶(GlcNAc-phosphotransferase,GNPT)作用,在 α-1,2 连接的甘露糖 C6-醇羟基上添加 N-乙酰葡萄糖胺-1-磷酸,形成 N-乙酰葡萄糖胺-1-磷酸-甘露糖的结构。随后在高尔基体反面膜囊(trans-Golgi-network,TGN)被第二个关键酶N-乙酰葡萄糖胺-1-磷酸二酯 α-N-乙酰葡萄糖胺糖苷酶(N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase,又称为 uncovering enzyme,UCE)水解去除 N-乙酰葡萄糖胺,即暴露出 M6P 结构[3],如图 1 所示。因此这两个关键酶对溶酶体代谢酶的转运起着重要的作用。

图1 溶酶体代谢酶甘露糖-6-磷酸的形成机制Figure 1 The biosynthetic mechanism of mannose-6-phosphate forming of lysosomal hydrolase
UCE 作为 M6P 形成过程中的第二个关键酶,是由 515 个氨基酸组成的 I 型膜蛋白,以四聚体形式存在,在高尔基体反面与细胞质膜之间循环[4-5]。UCE 编码基因NAGPA位于 16 号染色体上,含有 10 个外显子[5]。其 N-端 25 个氨基酸为信号肽,第 26~49 位为 24 个氨基酸的前体肽,第 50~447 位氨基酸为其核心区域,第 448~474 位氨基酸为 C-端的跨膜区,第 475~515 位氨基酸为 C-端胞浆尾肽[5]。在前体肽的 C-端含有蛋白酶 Furin 剪切位点——RARLPR↓D,定位于TGN 的蛋白酶 Furin 能够识别并剪切第 49 位精氨酸和第 50 位天冬氨酸之间的肽键,从而释放出成熟的 UCE。而未被剪切的 UCE 前体暂时没有活性,直到定位于 TGN 后才被 Furin 剪切产生活性[6]。
到目前为止,对于 UCE 的异源表达研究较少。Kornfeld 等[5]首次在 COS 细胞中表达并鉴定了 UCE。而 Zeng 等[7]在拟南芥种子中表达了具有活性的 UCE,并利用其对溶酶体代谢酶进行修饰。Do 等[6]分别在 CHO 细胞和昆虫细胞中进行了异源表达。大肠杆菌表达系统具有培养简单、快速、表达量高、成本低、易于工业放大等优势,本研究首次利用大肠杆菌表达系统进行 UCE 的表达和纯化,为大量制备 UCE 并在体外对溶酶体代谢酶进行糖基化修饰提供了可能。通过对不同长度的截短型 UCE 进行比较,并利用 UDP-Glo 试剂盒测定其活性,发现去掉信号肽、前体肽、跨膜区、胞浆尾肽,只留下核心区的 UCE 具有较高的活性。
1 材料与方法
1.1 材料
大肠杆菌Escherichia coliDH10b 作为质粒克隆的宿主菌,大肠杆菌 Shuffle T7 作为表达宿主菌均购自美国 NEB 公司;pCold-MBP 载体购自日本Takara 公司;酶活测定所需的 UDP-Glo 试剂盒购自美国 Promega 公司;底物 UDP-GlcNAc 购自美国 Sigma 公司。
1.2 方法
1.2.1 表达载体构建 构建截短不同结构肽段的突变体 UCE-1~UCE-5,分别用 UCE-MBP_BamH1-1/UCE_Sal1-1、UCE-MBP_BamH1-1/UCE_Sal1-2、UCE-MBP_BamH1-1/UCE_Sal1、UCE-MBP_BamH1-4/UCE_Sal1、UCE-MBP_BamH1/UCE_Sal1这 5 组引物对,以合成的全长 UCE 基因作为模板,扩增 5 种截短型的 UCE 基因片段,亚克隆到用限制性内切酶BamH I 和SalI 酶切处理的载体 pCold-MBP,即得到表达载体pCold-MBP-UCE-1/-2/-3/-4/-5。引物序列见表 1。

表1 PCR 扩增引物Table 1 PCR primers
1.2.2 载体转化 取适量质粒加入约 100 μl 的感受态细胞,轻弹管壁数次充分混匀,冰水浴 30 min,42 ℃ 热激 90 s,冰水浴 2 min;加入 800 μl LB 培养基(酵母提取物 5 g/L、胰蛋白胨 10 g/L、氯化钠 10 g/L,pH 7.0~7.2),37 ℃ 220 r/min 复壮培养 45 min,取适量菌液涂布到含有抗生素的 LB平板上,37 ℃ 培养。
1.2.3 菌株培养方法 将转化表达载体的重组菌挑取单菌落接种于 10 ml LB 培养基中,37 ℃220 r/min 培养 4 h 左右,按 1% 转接至 100 ml LB 培养基中,37 ℃ 220 r/min 培养至OD=0.4~0.6,15 ℃ 静置 30 min 后,加入 IPTG 诱导,15 ℃150 r/min 培养约 16 h。培养全程加入 100 μg/ml的氨苄青霉素。将发酵液离心弃上清,将沉淀用缓冲液重悬后超声波破碎细胞。超声波设置为最大功率的 39%,工作 3 s,暂停 3 s,超声破碎细胞5 min;大体积的菌体悬液采用高压匀浆破碎菌体(80 MPa,5 min);破碎后的样品 4 ℃、12000 r/min离心 10 min,分离上清和沉淀,再分别用 SDS-PAGE电泳检测。
1.2.4 酶活性测定 按照 UDP-Glo 试剂盒说明书的方法,配制 UDP 检测试剂。将纯化的 UCE与终浓度为 100 mmol/L 的 UDP-GlcNAc 在 25 μl体系中反应,反应缓冲液为 25 mol/L Tris-HCl,pH 7.5,150 mol/L NaCl,30 ℃ 200 r/min 反应 30 min。立即加入 25 μl UDP 检测试剂,30 ℃ 200 r/min 反应 1 h,立即在酶标仪中测定发光强度。
2 结果
2.1 不同长度的截短型 UCE 的表达与纯化
为了研究 UCE 的各个结构元件对其表达和活性的影响,本研究选择了 5 种不同长度的UCE突变体对其进行异源表达,分别为:①全长的UCE;②截去胞浆尾肽;③截去胞浆尾肽和跨膜区;④截去胞浆尾肽、跨膜区和信号肽;⑤截去胞浆尾肽、跨膜区、信号肽和前体肽,即只留下核心区。这 5 种 UCE 突变体分别表示为 UCE-1、UCE-2、UCE-3、UCE-4 和 UCE-5,其分子量分别为 56.1、51.4、48.6、45.9 和 43.2 kD(图 2)。

图2 截短形式 UCE 示意图Figure 2 Schematic of truncated forms of UCE
由于多种截短形式的 UCE 突变体含有疏水的跨膜区,可能会影响其成熟折叠和可溶性表达,故本研究在目的蛋白 UCE 的 N-端融合了一段约44 kD 大小的麦芽糖结合蛋白(maltose binding protein,MBP)序列[8],以期提高目的蛋白的可溶性。设计引物扩增不同长度的 UCE 基因片段,克隆到载体 pCold-MBP 上,即可得到融合 MBP 的不同截短型 UCE 突变体的大肠杆菌表达载体。在MBP 和 UCE 之间设计引入了 TEV 剪切位点,便于纯化后切去融合蛋白 MBP,释放出目的蛋白UCE。在 MBP 的上游含有 6 × His 纯化标签,便于利用 Ni2+离子亲和层析纯化目的蛋白。图 3 为表达载体示意图。

图3 截短型 UCE 表达载体的构建Figure 3 The construction of the truncated UCE expression vector
把含有 UCE 不同表达框的质粒转化到大肠杆菌 Shuffle T7 宿主细胞,对其诱导表达;将收集的菌体超声破碎后离心,分别对其上清部分和沉淀的细胞碎片部分进行 SDS-PAGE 电泳检测,结果如图 4 所示。转化 UCE-1/-2/-3/-4/-5 表达载体的宿主菌总蛋白的上清和沉淀部分均能够检测到目的蛋白 UCE 的融合表达,且从 UCE-1 到 UCE-5其分子量依次递减,与预期相符。而转化空载体的对照菌在诱导后仅能产生促溶标签蛋白 MBP 的条带。UCE-1~UCE-5 均在沉淀当中有大量的不溶性包涵体蛋白,除了全长 UCE-1 在上清中表达量较少以外,其余的突变体 UCE-2 到 UCE-5 均能在上清中有大量的可溶性融合表达。结果说明促溶标签 MBP 促进了融合蛋白的正确折叠并发挥其促溶作用。由于 UCE-1 的可溶表达较少,故仅对UCE-2~UCE-5 进行了亲和层析纯化。
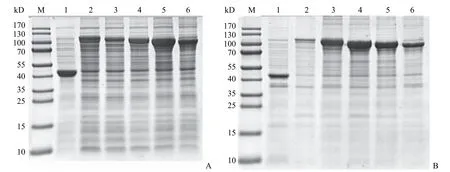
图4 表达 UCE 的菌体总蛋白沉淀(A)和上清(B)的 SDS-PAGE 分析Figure 4 SDS-PAGE analysis of insoluble (A) and supernatant (B) proteins of truncated UCE-expressing strains
按照 Ni2+离子亲和层析琼脂糖凝胶说明书进行操作,收集 300 mmol/L 咪唑洗脱液,对其进行SDS-PAGE 电泳分析,除含有少量的 40 kD 左右的 MBP 条带外,均只含有在 100 kD 左右的MBP-UCE 融合蛋白的单一条带(图 5)。

图5 纯化的融合 UCE 的 SDS-PAGE 分析Figure 5 SDS-PAGE analysis of recombinant fusion UCE
2.2 不同长度的截短型 UCE 突变体的活性测定
UCE 在溶酶体代谢酶的糖基化修饰过程中,能够水解 Man-P-GlcNAc 结构上的 GlcNAc,从而形成 M6P。由于 UCE 的天然底物为蛋白上的N-聚糖,蛋白酶解后的糖肽结构复杂难以检测分析,故本研究利用结构简单的底物 UDP-GlcNAc作为 UCE 的底物,UCE 能够水解其产生 UDP的特性,通过 UDP-Glo 试剂盒测定产生 UDP 的量,从而间接测定 UCE 的活性[7]。首先将底物UDP-GlcNAc 和酶反应,生成产物 UDP,再与试剂盒中提供的 UDP 检测试剂反应,该试剂能够通过一步反应将 UDP 转化成 ATP 并发光,测定产物的发光强度。
首先配制不同浓度的 UDP 溶液,利用该试剂盒测定发光强度,从而得到一条线性的 UDP 浓度-发光强度标准曲线,用来对 UDP 进行定量,如图 6A 所示。对于纯化的 UCE-2~UCE-5 融合蛋白,取 1.5 μg 蛋白和底物 UDP-GlcNAc 加入96 孔白板中反应,反应体系 25 μl,UDP-GlcNAc的终浓度为 100 μmol/L。反应结束后立即加入 25 μl配制好的 UDP 检测试剂,孵育 1 h 后在酶标仪中检测发光强度。如图 6B 所示,每微摩尔的 UCE-5能够产生较高的发光强度,且煮沸变性的样品酶活性降低,说明其具有较高的活性,而其余截短的UCE 活性则较低。UCE-5 的活性约为其他几种截短 UCE 活性的 2.5 倍。这说明前体肽的剪切对UCE 的活性非常重要,只有去掉前体肽才能使UCE 发挥活性,而去掉其信号肽、跨膜区和胞浆尾肽则不影响 UCE 的活性。

图6 UCE 酶活测定(利用 UDP-Glo 试剂盒进行 UCE 水解活性的分析,对照为反应缓冲液、反应缓冲液和底物以及变性失活的酶 UCE-2~UCE-5)(A:发光强度-UDP 浓度标准曲线;B:每微摩尔的酶催化所产生的发光强度)Figure 6 Determination of UCE activity using UDP-Glo kit [The negative control reactions were set up as buffer only,and buffer and substrate (without enzyme),and denatured enzymes UCE-2 to UCE-5,respectively] (A: Luminescence-UDP concentration standard curve;B: Luminescence of the UDP detection reactions with the truncated UCEs)
3 讨论
UCE 作为溶酶体代谢酶的糖基化修饰过程中的关键酶之一,具有重要的生理意义。由于 UCE是 I 型膜蛋白,C-端含有 27 个氨基酸的跨膜区,因此难以进行可溶性的异源表达。目前,仅有在哺乳动物细胞[5-6]、昆虫细胞[6]和植物[7]中通过截去跨膜区进行了异源表达和功能分析的报道。Zeng 等[7]利用拟南芥表达并在体外鉴定了 UCE,其中含有前体肽的形式,能够在植物体内被剪切,形成成熟的 UCE,并具有活性。该研究组还利用拟南芥共表达 UCE 与 GNPT,分别在体外和体内对治疗黏多糖贮积症 I 型的溶酶体水解酶艾杜糖醛酸酶(IDUA)进行糖基化修饰,得到了治疗效果更好的 IDUA。这说明植物体内含有类似 Furin 的蛋白酶,能够剪切前体肽,这对于 UCE 发挥活性至关重要。
由于天然的 UCE 是以前体形式合成的,需要经过蛋白酶 Furin 的剪切,去掉前体肽成为成熟酶才具有活性。而本研究中表达的多种形式的融合蛋白,分子量均在 100 kD 左右,且依次递减,这说明了大肠杆菌细胞中不存在类似 Furin 的剪切酶。因此,为了获得活性较高的 UCE,必须以截去前体肽的形式,即对本研究中的突变体 UCE-5 进行异源表达。该突变体不需要在体内经过蛋白酶加工,即可具有催化活性。而 UCE 能够水解UDP-GlcNAc,UDP-GlcNAc 又是高尔基体顺面上的 GNPT 的底物,作为添加 N-乙酰葡萄糖胺的糖基供体,过早地剪切 UCE 使其具有了活性,会提前降解 UDP-GlcNAc,从而影响 N-乙酰葡萄糖胺磷酸的生成。因此,UCE 只有被转运到高尔基体反面与 Furin 接触时才被剪切形成有活性的形式[6]。这也是 UCE 成熟机制需要经过前体肽剪切的原因。
本研究首次在大肠杆菌细胞中对 UCE 进行异源表达。利用 pCold-MBP 载体上的促溶标签蛋白 MBP 实现了除全长以外的其他突变体 UCE-2、UCE-3、UCE-4 和 UCE-5 的大量可溶性融合表达。因此利用该促溶标签蛋白 MBP 可以作为表达跨膜蛋白的一种策略。
由于 UCE 的天然底物是结构复杂的糖蛋白,经酶解处理的糖肽结构复杂难以直接获得。因此本研究选用结构简单且易得的底物 UDP-GlcNAc 作为 UCE 的底物,UCE 能够水解糖苷键,形成UDP 和 GlcNAc。再利用 UDP-Glo 试剂盒,可以间接测定产生 UDP 的生成量,从而反映出 UCE活性。利用该试剂盒可以避免文献报道同位素标记的方法[9],用 [3H]GlcNAc-α-Me-P-Man 作为底物,通过放射性测定的方法测定释放的 [3H]GlcNAc的量,使得 UCE 的酶活测定更加简便。
大肠杆菌表达系统作为一种简便快速的表达系统[10],可以作为 UCE 表达的候选宿主。由于目前大部分用于治疗溶酶体贮积症的溶酶体代谢酶都是由哺乳动物细胞生产,未经过糖基化改造,其形成的 M6P 含量往往较低,大大限制了其治疗效率[11]。因此可以通过大肠杆菌异源表达,大量获得M6P 合成的催化酶,建立体外的酶促生物催化体系,在体外对溶酶体代谢酶进行糖基化修饰,以提高其治疗效率[12]。但离体的生物催化合成溶酶体代谢酶 M6P 结构还需要与 GNPT 和甘露糖苷酶偶联催化,才能实现其糖基化修饰。本研究系统地进行了 UCE 的异源表达和功能活性分析,为建立体外生物催化合成溶酶体代谢酶的 M6P 结构提供了实验借鉴。
