一例PYGM基因复合杂合突变导致糖原累积症V型的诊断和基因检测分析
2022-11-29蒋琬姿徐昳文王依文朱肖诚龚颖芸周红文付真真
蒋琬姿,徐昳文,王依文,朱肖诚,龚颖芸,周红文,付真真
遗传资源
一例基因复合杂合突变导致糖原累积症V型的诊断和基因检测分析
蒋琬姿,徐昳文,王依文,朱肖诚,龚颖芸,周红文,付真真
南京医科大学第一附属医院内分泌科,南京 210029
糖原累积症V型是一种由肌糖原磷酸化酶(muscle glycogen phosphorylase,PYGM)缺陷引起的常染色体隐性遗传疾病,其临床特征为运动不耐受、再振作现象和血清肌酸激酶水平增高。本文报道了1例中国糖原累积症V型年轻男性患者,运动后双下肢无力、血肌酸激酶升高、肌肉磁共振可见下肢近端后群肌轻度脂肪浸润,基因检测结果显示先证者具有复合杂合型致病突变,分别为来自母亲的c.308T>C (p.L103P)变异和来自父亲的c.260_261delCT (p.S87Ffs*23)变异,其中前者为新发突变。本研究丰富了致病基因突变谱,有助于提高临床医生对糖原累积症V型的认识,并为该疾病的进一步遗传学研究提供参考。
糖原累积症V型;McArdle病;PYGM;罕见病
糖原累积症V型(glycogen storage disease type V, GSDV),又被称为McArdle病,是一种常染色体隐性遗传病[1],发病率约1/100,000~140,000[2]。1951年,英国McArdle医生最早发现一位30岁男性患者,他在运动后(包括咀嚼时)出现肌肉疼痛,剧烈运动后还出现无力和僵硬,但运动后血液中的乳酸没有增加,休息几分钟后症状可消失,McArdle医生推测该病的病因可能是运动后肌糖原无法分解,导致乳酸生成受损[3]。糖原磷酸化酶(glycogen phosphorylase,PYG)是催化糖原分解的限速酶,可促使细胞内糖原生成1-磷酸葡萄糖(glucose-1-phosphate, G1P)单体[4]。Schmid和Mahler等[5]于1959年确定了GSDV是由位于人染色体11q13上的肌糖原磷酸化酶(muscle glycogen phosphorylase,)基因突变引起该酶的功能缺失而导致的糖原分解障碍。
该病多发于15岁之前的儿童或青少年,也可在50岁后发病,男性多于女性[6]。患者表现为对运动不耐受、肌肉痉挛、疲劳和无力。在部分患者中,运动后可出现肌酸激酶急剧升高和横纹肌溶解伴肌红蛋白尿,严重的肌红蛋白尿可导致急性肾功能衰竭。在许多McArdle病患者中观察到以下现象[6~8]:在运动5 min左右患者出现肌痛、疲劳,经过短暂休息几分钟或减慢运动后,症状突然消失且对运动的耐受性有所提高,这种现象被定义为再振作现象(second wind phenomenon),又称继减现象。在患者成年后,随着脂肪的替代,肌肉持续无力和萎缩[9]。McArdle病大部分患者病情较轻,不会影响患者寿命[10],但病情较重者可能因肌红蛋白尿而导致肾功能衰竭、出现全身性肌无力甚至进行性呼吸衰竭危及生命[10]。束臂运动试验比较患者运动前后血乳酸水平可辅助诊断GSDV,正常人运动后血乳酸水平往往增高,患者因为缺乏PYGM,肌糖原不能分解,运动后血乳酸不升高。本文主要报道了1例McArdle病年轻男性患者,基因检测分析发现其具有基因复合杂合突变,分别为来自母亲的c.308T>C (p.L103P)变异和来自父亲的c.260_261delCT (p.S87Ffs*23)变异,其中前者为新发突变。本研究丰富了致病基因突变谱,为该疾病的遗传学研究提供了新的临床资源,同时也有助于糖原累积症V型的早期诊断和加深临床医生对基因的了解。
1 对象与方法
1.1 对象及临床资料收集
患者,男,23岁,因“运动后双下肢无力伴酸痛20年”于2019年至我院就诊,收集患者的病史、体格检查、实验室检查、影像学检查等临床资料。本研究获得南京医科大学第一附属医院医学伦理委员会批准,患者及其父母均知情同意。
1.2 全外显子测序及家系验证
患者及其父母留取2 mL全血并委托北京金准基因科技有限公司进行DNA基因组提取,通过目标区域捕获技术对富集的基因利用高通量测序仪对患者进行全外显子测序分析,测序完成后利用BWA软件将过滤后的序列比对到NCBI数据库人类基因组参考序列(hg19)上,随后使用北京金准基因科技有限公司自有的本地数据库和分析软件进行进一步注释和筛选。对患者父母进行了针对所检出变异位点的Sanger测序以验证全外显子测序所发现的基因变异并确认变异位点的父母来源。
1.3 新突变位点致病性预测与蛋白质二级结构预测
使用Polyphen2、SIFT、Mutation Taster等工具对位点的致病性进行预测;利用DNAMAN软件(Lynnon Biosoft)对不同物种的基因突变位点进行保守性分析。使用AlphaFold(https://alphafold. ebi.ac.uk)和SWISSMODEL蛋白质结构建模工具(https://swissmodel.expasy.org)预测PYGM蛋白的二级结构。同时,根据美国医学遗传学会(American College of Medical Genetics, ACMG)遗传变异分类标准将检测到的变异分类为“致病”、“可能致病”和“意义不明确”。
2 结果与分析
2.1 患者的临床表现
患者,男,23岁,因“运动后双下肢无力伴酸痛20年”于2019年至我院就诊,患者剖腹产出生,出生时体重及先天性反射正常,母乳喂养,从小食欲一般,比女生跑步慢,体育成绩不佳,跑步350 m后感双下肢无力酸痛,休息3~5 min即症状消失,可继续运动;起跳正常,立定跳远尚可,做仰卧起坐5个之后腹部酸胀抽痛明显,休息后好转。随着年龄增长,患者症状未见明显好转,病程中未出现酱油尿,父母非近亲婚配,家族中无类似患者。查体:身高:161 cm,体重:70 kg,四肢无肌肉萎缩和肥大,肌力肌张力正常,Martinuzzi指数[11]分级为I级(表1)。生化全套分析:丙氨酸氨基转移酶70.0 ↑ U/L(正常参考值:9~50 U/L),天门冬氨酸氨基转移酶61.6 ↑ U/L (正常参考值:15~40 U/L),乳酸脱氢酶236 U/L (正常参考值:140~271 U/L),肌酸激酶5036 ↑ U/L (正常参考值:0~171 U/L),尿酸503 ↑ μmol/L (正常参考值:208~428 U/L),血清肌钙蛋白I阴性,高敏肌钙蛋白T 32.96 ↑ ng/L(正常参考值:0~14 ng/L),肌酸激酶同工酶53.3 ↑ U/L (正常参考值:0~25 U/L),肌红蛋白151 ↑ μg/L(正常参考值:0~46 μg/L),血常规、血脂、血糖、免疫五项、抗核抗体、抗ENA抗体、心电图、超声心动图、肝胆胰脾双肾输尿管及甲状腺彩超、头颅MRI平扫及肌电图均未见明显异常,下肢近端肌肉MRI可见大腿后群部分轻度脂肪浸润(图1)。

表1 Martinuzzi指数评估McArdle病严重程度
2.2 基因检测结果分析
全外显子组测序报告显示该患者为c.260_261delCT(p.S87Ffs*23)及c.308T>C(p.L103P)复合杂合突变,Sanger测序进一步确认了上述突变,前者为已知突变位点(来自父亲),后者为新发突变位点(来自母亲)(图2A),可诊断为GSDV。
该患者为复合杂合突变,第一个来源于父亲的突变位点为c.260_261delCT(p.S87Ffs*23),260~261位CT碱基缺失突变导致氨基酸移码突变并在23位后终止,仅有1例病例报道;第二个来源于母亲的突变位点为c.308T>C(p.L103P),308位T碱基突变为C碱基导致编码氨基酸由亮氨酸变为脯氨酸,在Clinvar(https://www.ncbi.nlm.nih.gov/clinvar),LOVD数据库(https://databases.lovd.nl/shared/genes),gnomAD (http://gnomad.broadinstitute.org/about),1000 Genomes(http://browser.1000genomes.org/)检索中均未见报道。经过生物信息学预测c.260_ 261delCT (p.S87Ffs*23)能够改变PYGM蛋白的二级结构,但c.308T>C (p.L103P)似乎对PYGM蛋白的二级结构影响不大(图2B)。该患者基因两个突变位点氨基酸在不同物种中高度保守(图2C)。根据美国医学遗传学会(American College of Medical Genetics, ACMG)制定的序列变异解读标准和指南判定c.260_261delCT (p.S87Ffs*23)变异为可能致病突变,c.308T>C (p.L103P)变异为意义不明确突变,但依据SIFT (http://sift.jcvi.org/)、Polyphen2 (http://genetics.bwh.harvard.edu/pph2)及Mutation Taster (http://www.mutationtaster.org/)等蛋白损伤预测软件评估c.308T>C (p.L103P)突变极有可能致病(表2)。

图1 患者下肢肌肉可见近端后群肌肉轻度脂肪浸润
A:肌肉磁共振T1序列图;B:肌肉磁共振T1-FS序列图。

图2 患者及患者父母的基因检测结果分析
A:基因复合杂合突变,c.260_261delCT (p.S87Ffs*23),c.308T>C (p.L103P),前者来自父亲,后者来自母亲。B:PYGM蛋白二级结构预测。黑色箭头指向的红色位置表示PYGM p.L103P 氨基酸突变位置。C:基因p.S87位点及p.L103位点基因突变位点保守性分析。
3 讨论
该患者从小存在运动不耐受,包括跑步慢、肌肉疲劳、肌痛等,但当患者休息几分钟或减少运动强度后,便会出现再振作现象,能够在症状改善的情况下继续进行运动,但目前未出现“酱油色尿”(肌红蛋白尿)发作。患者运动后血清肌酸激酶急剧升高,肌酸激酶同工酶、肌红蛋白及尿酸均升高,符合典型GSDV临床表现,同时在基因检测上明确了基因突变,因此该患者可明确诊断为GSDV。
GSDV也称为McArdle病,是肌糖原病中最常见的一类疾病[12]。McArdle病可以通过基因检测发现最常见的突变或肌肉活检或分子遗传学检测做出诊断[13]。在Clinvar检索共有802个已知突变位点,在西班牙等欧美国家人群中最普遍的突变是p.R50X、p.W798R和p.G205S,约占所有突变的61%,其中p.R50X是最普遍的[14]。有研究发现基因单核苷酸多态位点突变也与肌肉有氧运动能力有关。rs490980位点不同基因型的运动员有氧运动能力差异明显,CC型显著低于TT型和TC型,由此推测rs490980位点多态性可能对mRNA的表达有一定影响,从而影响了代谢过程[15]。而我国GSDV病例报道很少,已有的文献报道见表3。

表2 PYGM基因未报道新突变位点致病性预测
肌肉收缩所需的能量由三磷酸腺苷(ATP)提供,而糖酵解是肌肉产生ATP的必需来源。但是,在McArdle病中,肌糖原磷酸化酶缺乏会导致糖原分解受阻,并导致ATP生成减少同时抑制三羧酸循环。因此,用于骨骼肌收缩的ATP不足,导致运动不耐受、运动中肌肉疼痛、痉挛、乏力和肌酸激酶升高[25]。在McArdle病中,高尿酸血症及痛风的报道较少[26],而该患者出现了高尿酸血症,有研究认为,由于糖原分解代谢障碍,患者加剧了ATP消耗,两个二磷酸二腺苷(ADP)分子可以结合,通过腺苷酸激酶再生ATP以尝试跟上ATP需求,反应中将产生副产物单磷酸腺苷(AMP),并通过AMP脱氨酶代谢产生肌苷单磷酸酯(IMP)和氨(NH3)。IMP通过黄嘌呤氧化酶代谢为肌苷,然后代谢为次黄嘌呤及黄嘌呤,经黄嘌呤氧化酶催化将次黄嘌呤和黄嘌呤还原为尿酸,因此McArdle病中出现高尿酸血症可能为肌源性高尿酸血症(图3)[27]。
GSDV尚无特定的治疗方法,目前该疾病的主要治疗方案是对症支持治疗并避免剧烈运动,因此早期诊断对促进生活方式的改变很重要。
饮食上,有研究表明GSDV患者运动前摄入葡萄糖可提高运动耐力,并可预防运动引起的横纹肌溶解[28]。Andersen等[29]在随机交叉研究设计中比较了富含碳水化合物(20%脂肪、15%蛋白质和65%碳水化合物)和富含蛋白质(55%蛋白质,30%碳水化合物和15%脂肪)组成的饮食,通过进行自行车骑行试验,发现富含碳水化合物的饮食使患者平均最大摄氧量、心率显著下降。2017年,第211届ENMC国际研讨会对McArdle病建议患者每日摄入10%~15%的蛋白质,<35%的脂肪和>50%的碳水化合物[14]。
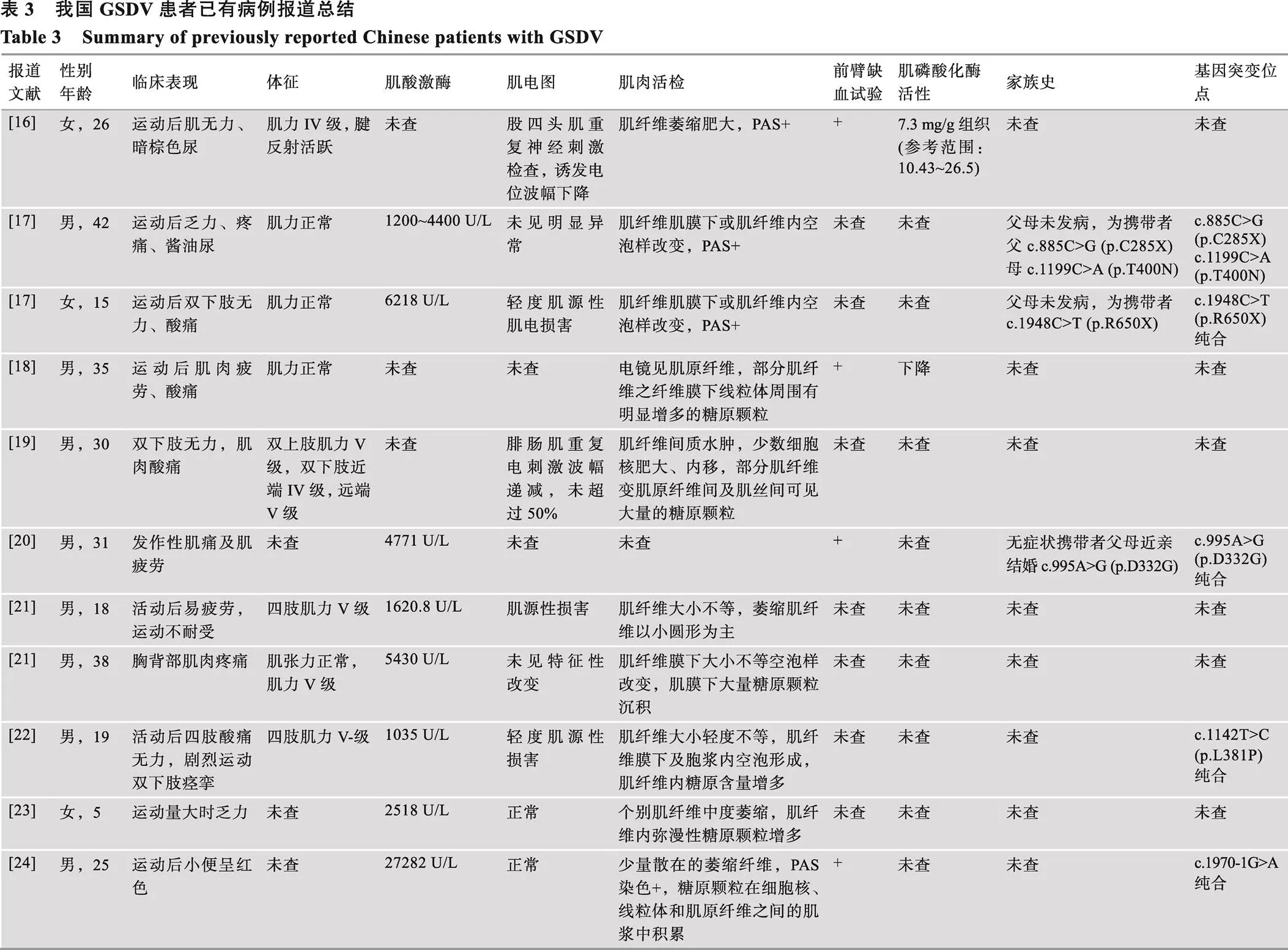
图3 GSDV发病机制及其导致高尿酸血症表型的可能机制
Fig. 3 The pathogenesis of GSDV and its possible mechanism leading to hyperuricemia
定期的有氧运动(结合运动前的碳水化合物摄入)有助于葡萄糖氧化转变为运动的替代能源(例如脂肪酸),从而提高运动耐力。Haller等[30]连续14周对8例患者使用自行车测功机(每周4次,运动强度相当于其最大心率的60%~70%,运动时间为30~40 min/次)训练,Matez-Munoz等[31]对9名GSDV患者实施8个月训练(每周步行5次或自行车测功机训练5次,强度和疗程持续时间与Haller研究类似),研究结果均显示有氧运动训练对峰值工作能力、最大有氧运动量、心输出量的显著改善。因此适当的有氧运动对改善GSDV患者运动耐力是有效的。
维生素B6是参与氨基酸代谢和神经递质生物合成的众多酶的辅因子。肌糖原磷酸化酶也需要维生素B6作为辅因子[32],对症应用维生素B6(60~ 90 mg/天)可以改善突变型磷酸化酶的活性和骨骼肌的无氧酵解并增强其在患者骨骼肌中的残留活性[33,34]。在先前McArdle病患者的报告中,补充维生素B6数周后肌肉疲劳显著降低[32]。也有研究认为,运动后磷酸肌酸会迅速消耗,补充低剂量肌酸(每天60 mg/kg)可能也是有益的[35]。
综上所述,本文报道了1例基因突变导致McArdle病以及基因1个新发突变位点。对于McArdle病等罕见病,临床表现缺乏特异性,很多症状难以与其他疾病鉴别,基因检查能明确诊断,并分析病变基因的来源。本研究丰富了基因突变谱,有助于提高临床医生对McArdle疾病的临床诊治和致病机制的掌握。
[1] Santalla A, Nogales-Gadea G, Encinar AB, Vieitez I, González-Quintana A, Serrano-Lorenzo P, Consuegra IG, Asensio S, Ballester-Lopez A, Pintos-Morell G, Coll- CantíJ, Pareja-Galeano H, Díez-Bermejo J, Pérez M, Andreu AL, Pinós T, Arenas J, Martín MA, Lucia A. Genotypic and phenotypic features of all Spanish patients with McArdle disease: a 2016 update. BMC Genomics, 2017, 18(Suppl 8): 819.
[2] De Castro M, Johnston J, Biesecker L. Determining the prevalence of McArdle disease from gene frequency by analysis of next-generation sequencing data. Genet Med, 2015, 17(12): 1002–1006.
[3] McARDLE B. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci, 1951, 10(1): 13–35.
[4] Lebo RV, Anderson LA, DiMauro S, Lynch E, Hwang P, Fletterick R. Rare McArdle disease locus polymorphic site on 11q13 contains CpG sequence., 1990, 86(1): 17–24.
[5] Schmid R, Mahler R. Chronic progressive myopathy with myoglobinuria: demonstration of a glycogenolytic defect in the muscle., 1959, 38: 2044–2258.
[6] 中华医学会神经病学分会, 中华医学会神经病学分会神经肌肉病学组, 中华医学会神经病学分会肌电图与临床神经生理学组. 中国糖原累积性肌病诊治指南. 中华神经科杂志, 2016, 49(1): 8–16.
[7] Braakhekke JP, de Bruin MI, Stegeman DF, Wevers RA, Binkhorst RA, Joosten EM. The second wind phenomenon in McArdle's disease., 1986, 109 (Pt 6): 1087–1101.
[8] Haller RG, Vissing J. Spontaneous "second wind" and glucose-induced second "second wind" in McArdle disease: oxidative mechanisms., 2002, 59(9): 1395–1402.
[9] Witting N, Duno M, Piraud M, Vissing J. Severe axial myopathy in McArdle disease., 2014, 71(1): 88–90.
[10] Quinlivan R, Buckley J, James M, Twist A, Ball S, Duno M, Vissing J, Bruno C, Cassandrini D, Roberts M, Winer J, Rose M, Sewry C. McArdle disease: a clinical review., 2010, 81(11): 1182–1188.
[11] Martinuzzi A, Sartori E, Fanin M, Nascimbeni A, Valente L, Angelini C, Siciliano G, Mongini T, Tonin P, Tomelleri G, Toscano A, Merlini L, Bindoff LA, Bertelli S. Phenotype modulators in myophosphorylase deficiency., 2003, 53(4): 497–502.
[12] Nogales-Gadea G, Santalla A, Brull A, de Luna N, Lucia A, Pinós T. The pathogenomics of McArdle disease--genes, enzymes, models, and therapeutic implications., 2015, 38(2): 221–230.
[13] Rubio JC, Garcia-Consuegra I, Nogales-Gadea G, Blazquez A, Cabello A, Lucia A, Andreu AL, Arenas J, Martin MA. A proposed molecular diagnostic flowchart for myophosphorylase deficiency (McArdle disease) in blood samples from Spanish patients., 2007, 28(2): 203–204.
[14] Quinlivan R, Andreu AL, Marti R, Workshop P. 211th ENMC International Workshop: development of diagnostic criteria and management strategies for McArdle disease and related rare glycogenolytic disorders to improve standards of care. 17–19 April 2015, Naarden, the Netherlands., 2017, 27(12): 1143– 1151.
[15] Wu J, Hu Y, Li CY, Yi LY. Association study of PYGM gene and haplotypes with women endurance athletes physiological phenotypes., 2014, 26(4): 376–379.
吴剑, 胡扬, 李燕春, 衣龙彦. PYGM基因SNP及单体型多态性与女子长跑运动员生理表型的关联性. 首都体育学院学报, 2014, 26(4): 376–379.
[16] 沈定国. McArdle氏病的不常见类型一例报告. 中华神经精神科杂志, 1985, 18(3): 184.
[17] 岳冬曰, 高名士, 罗苏珊, 刘华, 李颖, 易芳芳, 赵重波, 毕海霞. McArdle病二例. 中华神经科杂志, 2017, 50(1): 56–58.
[18] 沙松林, 戴蔼善, 王宗根, 倪赞明, 邱传禄, 钟学礼, 朱世能. 肌糖原累积病一例报告. 中华内科杂志,1981, 20(9): 563–564.
[19] 刘志军, 张小荣, 杨瑞龙, 李妍怡. 早期McArdle病1例报告. 中风与神经疾病杂志, 2014, 31(2): 179–180.
[20] Xie RR, Yang YB, Jin P. Identification of a novel PYGM mutation in a McArdle disease patient misdiagnosed as hypokalemic periodic paralysis., 2020, 43(5): 697–698.
[21] 刘华旭. 糖原累积性肌病的基因特征、自噬及蛋白质组学研究[学位论文]. 中国人民解放军医学院, 2018.
[22] 张睿. 糖原累积病2例临床与基因分析[学位论文]. 山东大学, 2016.
[23] 代英杰. 糖原累积病临床病理分析[学位论文]. 中国协和医科大学, 2009.
[24] Lau NKC, Chong YK, Cheung KPK, Loo KT, Ching CK. McArdle disease presenting as abnormal liver function: biochemical, anatomical and genetic characterisation in the first genetically confirmed Chinese family with a novel splicing variant., 2021, 53(5): 670–673.
[25] Satoh A, Hirashio S, Arima T, Yamada Y, Irifuku T, Ishibashi H, Motoda A, Sueda Y, Masaki T. Novel Asp511Thr mutation in McArdle disease with acute kidney injury caused by rhabdomyolysis., 2019, 8(3): 194–199.
[26] Üsküdar Cansu D, Erdoğan B, Korkmaz C. Can hyperuricemia predict glycogen storage disease (McArdle's disease) in rheumatology practice? (Myogenic hyperuricemia)., 2019, 38(10): 2941–2948.
[27] Kitaoka Y, Ogborn DI, Nilsson MI, Mocellin NJ, MacNeil LG, Tarnopolsky MA. Oxidative stress and Nrf2 signaling in McArdle disease., 2013, 110(3): 297–302.
[28] Andersen ST, Haller RG, Vissing J. Effect of oral sucrose shortly before exercise on work capacity in McArdle disease., 2008, 65(6): 786–789.
[29] Andersen ST, Vissing J. Carbohydrate- and protein-rich diets in McArdle disease: effects on exercise capacity., 2008, 79(12): 1359–1363.
[30] Haller RG, Wyrick P, Taivassalo T, Vissing J. Aerobic conditioning: an effective therapy in McArdle's disease., 2006, 59(6): 922–928.
[31] Maté-Muñoz JL, Moran M, Pérez M, Chamorro-Viña C, Gómez-Gallego F, Santiago C, Chicharro L, Foster C, Nogales-Gadea G, Rubio JC, Andreu AL, Martín MA, Arenas J, Lucia A. Favorable responses to acute and chronic exercise in McArdle patients., 2007, 17(4): 297–303.
[32] Phoenix J, Hopkins P, Bartram C, Beynon RJ, Quinlivan RC, Edwards RH. Effect of vitamin B6 supplementation in McArdle's disease: a strategic case study., 1998, 8(3–4): 210–212.
[33] McCormick Z, Chu SK, Chang-Chien GC, Joseph P. Acute pain control challenges with buprenorphine/naloxone therapy in a patient with compartment syndrome secondary to McArdle's disease: a case report and review., 2013, 14(8): 1187–1191.
[34] Sato S, Ohi T, Nishino I, Sugie H. Confirmation of the efficacy of vitamin B6 supplementation for McArdle disease by follow-up muscle biopsy., 2012, 45(3): 436–440.
[35] Vorgerd M, Grehl T, Jager M, Muller K, Freitag G, Patzold T, Bruns N, Fabian K, Tegenthoff M, Mortier W, Luttmann A, Zange J, Malin JP. Creatine therapy in myophosphorylase deficiency (McArdle disease): a placebo-controlled crossover trial., 2000, 57(7): 956–963.
Diagnosis and genetic analysis of a case with glycogen storage disease type V caused by compound heterozygous mutations in thegene
Wanzi Jiang, Yiwen Xu, Yiwen Wang, Xiaocheng Zhu, Yingyun Gong, Hongwen Zhou, Zhenzhen Fu
Glycogen storage disease type V is an autosomal recessive genetic disorder caused by muscle glycogen phosphorylase (PYGM) deficiency, which is characterized by exercise intolerance, second wind phenomena and high level of serum creatine kinase. In this study, we reported a Chinese young man with glycogen storage disease type V, with lower extremity weakness after exercise, increased creatine kinase, and slight fat infiltration in the posterior group of thigh muscle by magnetic resonance imaging (MRI). The proband had complex heterozygousdisease-causing mutations, including c.308T>C (p.L103P) variant transmitted from the mother and c.260_261delCT (p.S87Ffs*23) from the father, of which the former was a novelmutation. This study enriched thepathogenic gene mutation spectrum, contributed to improve clinicians' understanding of glycogen storage disease type V and provided a reference for further genetic study of the disease.
glycogen storage disease type V; McArdle disease; PYGM; rare disease
2022-06-30;
2022-09-07;
2022-09-16
国家重点研发计划(编号:2019YFA0802701,2018YFA0506904),国家自然科学基金面上项目(编号:82170882)和国家自然科学基金重大研究计划培育项目(编号:91854122)资助[Supported by the National Key Research and Development Program of China (Nos. 2019YFA0802701, 2018YFA0506904), the National Natural Science Foundation of China (No. 82170882) and the Major Research Plan of the National Natural Science Foundation of China (No. 91854122)]
蒋琬姿,在读博士研究生,专业方向:肥胖、脂代谢和罕见代谢病。E-mail: 1995jwz@sina.com
付真真,博士,主治医师,研究方向:肥胖、脂代谢和罕见代谢病。E-mail: zhenzhen1127@foxmail.com
10.16288/j.yczz.22-223
(责任编委: 孟卓贤)
