藜麦基因组学与重要农艺性状位点研究进展
2022-11-29李玲红苟彤任爱霞丁鹏程林文武祥云孙敏高志强
李玲红,苟彤,任爱霞,丁鹏程,林文,武祥云,孙敏,高志强
综 述
藜麦基因组学与重要农艺性状位点研究进展
李玲红1,苟彤1,任爱霞1,丁鹏程1,林文1,武祥云2,孙敏1,高志强1
1. 山西农业大学农学院,太谷 030801 2. 山西稼琪农业科技有限公司,太原 030006
藜麦(Willd.)作为20世纪新兴的健康食物,因其营养成分全面、抗逆性强等特性备受关注,在国际上享有“营养黄金”、“素食之王”、“未来食品”的美誉。近年来随着基因组学和高通量测序技术的快速发展,藜麦高质量的全基因组序列得以完成并开展了系列关键基因功能研究。本文总结了藜麦基因组学、重要转录因子基因家族分析、遗传图谱构建和重要性状QTL定位和重要农艺和产量性状基因的研究进展。此外,针对目前藜麦育种的现状,本文还提出了藜麦育种存在的5个关键问题,并指出了未来藜麦遗传改良和育种的4个重要方向,旨在为实现未来藜麦的定向遗传改良提供参考。
藜麦;基因组学;遗传育种;性状;基因
藜麦(, Willd.)属于藜科藜属,其种子营养丰富,蛋白质含量高,是联合国粮食及农业组织(FAO)唯一认定的一种单体作物(即可满足人类全部基本营养需求的食物),是一种极具潜力的世界性粮食作物,因此2013年被联合国定为藜麦年(Year of Quinoa)[1]。藜麦起源于南美洲的安第斯山脉,生态适应能力强,具有较强的耐寒、耐旱、耐瘠薄、耐盐碱等抵抗多种非生物逆境的能力,是研究植物应对高盐度和干旱的新型模式作物。
尽管藜麦在国际上的重要性日益增加,但藜麦产业在我国的发展刚刚起步,有很多亟待解决的问题。例如在栽培育种方面,存在优异种质资源少、优良品种缺乏、配套栽培技术不完善等问题,需要藜麦工作者大力开展引种工作,加快培育优质、高产、广适的藜麦品种,加强配套高产栽培技术研究,同时加快绿色有机食品认证[2,3]。随着藜麦种植面积的不断增长,系统研究其遗传组成、基因型和环境的互作及其营养特性的遗传基础等也变得愈加重要。
本文总结了近年来藜麦基因组和遗传育种的研究成果,并在此基础上提出了目前藜麦育种改良工作中存在的关键问题,对藜麦未来遗传育种改良和发展方向进行了探讨,旨在为藜麦的未来遗传改良提供重要参考。
1 藜麦基因组研究进展
藜麦是异源四倍体,由祖先二倍体A基因组()和B基因组()杂交而成,基因组的复杂性在一定程度上限制了藜麦基因组学的发展[4]。随着高通量测序技术的快速发展,涌现出大量的藜麦序列信息,藜麦基因组学的研究为藜麦遗传多样性分析、功能基因挖掘及基因功能的研究及各类组学的分析等奠定了重要基础。2016年,藜麦全基因组测序及组装完成,相关研究成果发表于[5]。该研究利用Illumina Hiseq 2500结合PacBio RS II的测序方法,最终组装得到近25,000个scaffold,N50为86K。该研究主要完成基因组结构研究、同科物种基因组的基因家族分析以及抗非生物胁迫信号通路的基因进化分析等,但由于进化部分的分析过于简单,不能完全解析四倍体基因组的进化问题。2017年,Jarvis等[6]在上公布了高质量的藜麦全基因组序列,该研究利用单分子测序、光学图谱及遗传图的方法完成了全基因组测序及基因组组装,并利用二代测序方法组装了两个祖先二倍体;同时,还对22个异源四倍体藜麦完成重测序;此外,该研究确定了藜麦的进化地位并找到了控制种子中合成皂苷的重要转录因子。同年,Wang等[7]完成了藜麦叶绿体基因组测序工作。藜麦cpDNA (chloroplastic DNA)全长为151,169 bp,包含120种基因,其中蛋白编码基因87种、tRNA 29种、rRNA 4种,总GC含量为37.3%。
除此之外,其他藜麦基因组资源包括几个转录组测序数据库(EST或RNA-seq)和细菌人工染色体(BAC)文库也得以公布[8, 9],这些数据丰富了藜麦的基因组序列信息,对于藜麦转录因子基因家族的鉴定、连锁图谱的构建及功能基因/QTL的挖掘及功能分析等提供了极为有利的条件{Raney, 2014 #15;Stevens, 2006 #13}。
2 藜麦重要转录因子基因家族研究进展
转录因子是生物中重要的调控因子,广泛参与生物生长、发育、代谢及环境响应的调控过程[10]。结合藜麦全基因组测序信息,利用生物信息学方法,对藜麦转录因子进行全基因组鉴定,并对其理化性质、系统发育、基因结构、保守结构域、保守基序、组织表达和抗逆胁迫的表达进行分析,目前在藜麦中已有10余种转录因子基因家族被相继报道,包括NAC[11](NAM、ATAF和CUC首字母缩写)、MADS-box[12](MCM1、AG、DEF和SRF的首字母缩写)、WRKY[13]、TH[14](trihelix)、NHX[15](Na+/H+逆向转运蛋白)、TCP[16](TB1、CYC和PCF1/2首字母缩写)、SPL[17](SQUAMOSA-promoter binding protein-like)、Hsf[18](heat shock transcription factors)、GRF[19](growth- regulating factor)、WOX[20](WUSCHEL-related homeobox)、KEA[21](K+efflux antiporter)、FAX[22](fatty acid export)和NLP[23](NIN-like protein)等。各家族的名称、主要功能、家族成员数量、基因组织表达特异性及基因功能预测详见表1。
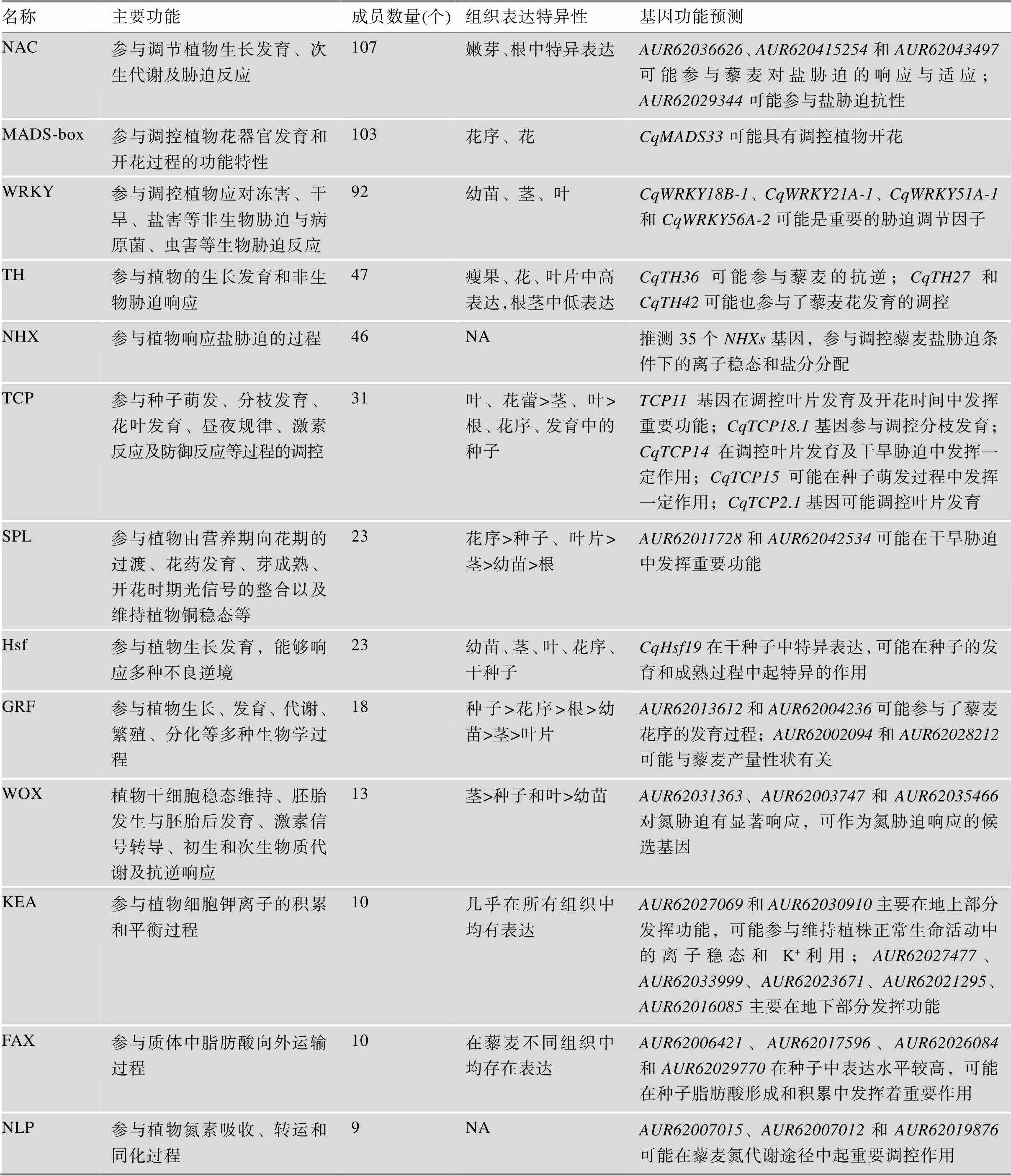
表1 藜麦转录因子基因家族的鉴定及表达分析
表中数据引自参考文献[11~23];NA:未找到任何部位的表达数据。
3 藜麦连锁图谱构建及重要性状的基因定位
3.1 藜麦分子标记的开发和连锁图谱的构建
遗传标记是现代育种技术的重要组成部分,使藜麦基因型的改良成为可能[24]。1993年,Fairbanks等[25]首次报道了利用随机扩增多态性DNA (random amplified polymorphic DNA, RAPD)标记的藜麦DNA标记,在16份藜麦材料中发现了26个多态性标记。2004年,扩增片段长度多态性分子标记(amplified fragment length polymorphism, AFLP)也在藜麦中得以开发并利用[26]。2005年,利用31份藜麦种质资源开发了第一套简单序列重复(simple sequence repeat, SSR)标记,包括208个SSR多态性标记[27]。随后,单核苷酸多态性(single-nucleotide polymorphisms, SNPs)标记也应用于藜麦分子标记的检测及遗传多样性的分析[24]。2017年,通过对11份藜麦种质资源进行重新测序,从基因组InDel变异中,新开发并验证了85个多态性InDel标记。结合已有的62个SSR标记,该研究共用147个标记对129份藜麦材料进行基因分型,结果表明藜麦基因组类型多样[28]。
Maughan等[26]利用智利低海拔藜麦品种KU-2与秘鲁高原藜麦品种0654杂交获得的80个F2个体,构建了藜麦的第一个遗传连锁图谱,该图谱包括255个多态性标记(AFLPs、RAPDs和SSRs),构建了35个连锁群,总遗传距离为1020 cM,每个标记的平均密度为4.0 cM。第二个藜麦基因连锁图谱是利用KU-2与0654品种杂交产生的82个重组自交系(recombinant inbred lines, RIL)群体构建,该图谱包含200个SSR标记和70个AFLP标记,构建了38个连锁群,遗传距离共913 cM[29]。此后,利用两个具有共同父本(0654高原藜麦品种)包含128个重组自交系的高代RIL群体,对451个多态性SNP标记进行连锁分析,得到29个遗传连锁群,连锁长度为1404 cM,平均每个连锁群3.1 cM。得益于更大作图群体的基因分型和更高的标记密度,这张图谱的完整性接近预测的藜麦总长度(1700 cM)[24]。最新的一份连锁图谱由Jarvis等[6]将前人的图谱与两份新的连锁图谱相结合,最终的连锁图谱包含了横跨2034 cM的18个连锁群上的6403个标记。综上所述,藜麦中已开发的分子标记和构建的连锁图谱相较于其他作物还是相对较少的,未来的研究需要构建更多更紧密的遗传图谱,用于定位更多有价值的基因或QTL。
3.2 藜麦中已经定位的基因/QTL
迄今为止,在藜麦中定位了一些重要农艺性状的基因/QTL,具体信息详见表2和表3。Cervantes和van Loo[30]利用“Red Carina”(种子苦, 颜色深)和“Atlas”(非苦的欧盟品种)杂交产生的94个F3个体,构建了包含1076个SNP的QTL连锁图谱,定位了花色、开花时间和产量等22个农艺性状相关的QTL。此外,2020年Sarange等[31]通过对310份材料进行全基因组测序,发现了290万个高信度多态性SNP位点,全基因组关联研究发现600个SNP与17个农艺性状稳定相关。其中与千粒重相关的候选基因有2个,与抗霜霉病相关的候选基因有一个类似的抗性基因。此外该研究鉴定了4个农艺性状的多效性作用位点,这些位点对光周期响应高,因此对不同环境的适应具有重要意义。最近,Maldonado等[32]利用亲本PI614889 (智利品种)与CHEN-109 (秘鲁品种)杂交构建的F2分离群体构建了高密度遗传图谱,结合2个分离世代(F2和F3)的表型数据定位了15个QTL,包括开花日数、成熟日数、株高、穗长、千粒重、皂苷含量、霉变敏感性等性状。同时,该研究在一些多效QTL中发现了多个与光周期响应和开花时间调控相关的候选基因。综上所述,藜麦中已定位的重要农艺性状基因/QTL是有限的,且定位区间都比较大,需要进一步精细定位,找到区间内的候选基因,进行基因的克隆和功能验证,从而更好地服务于藜麦的遗传改良育种。

表2 藜麦中已定位的基因汇总信息
表中数据引自参考文献[31]。

表3 藜麦中已定位的QTL汇总信息
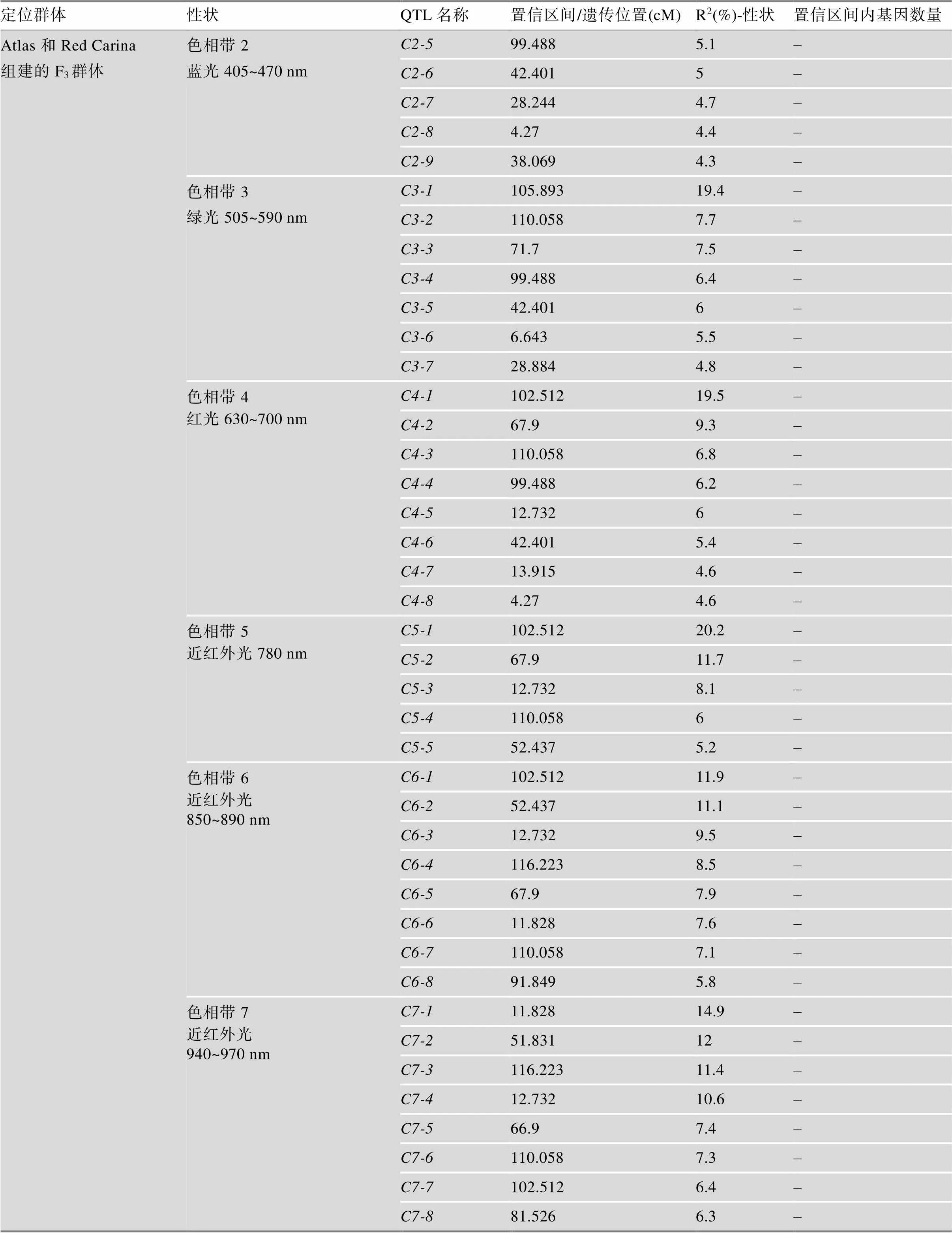
续表
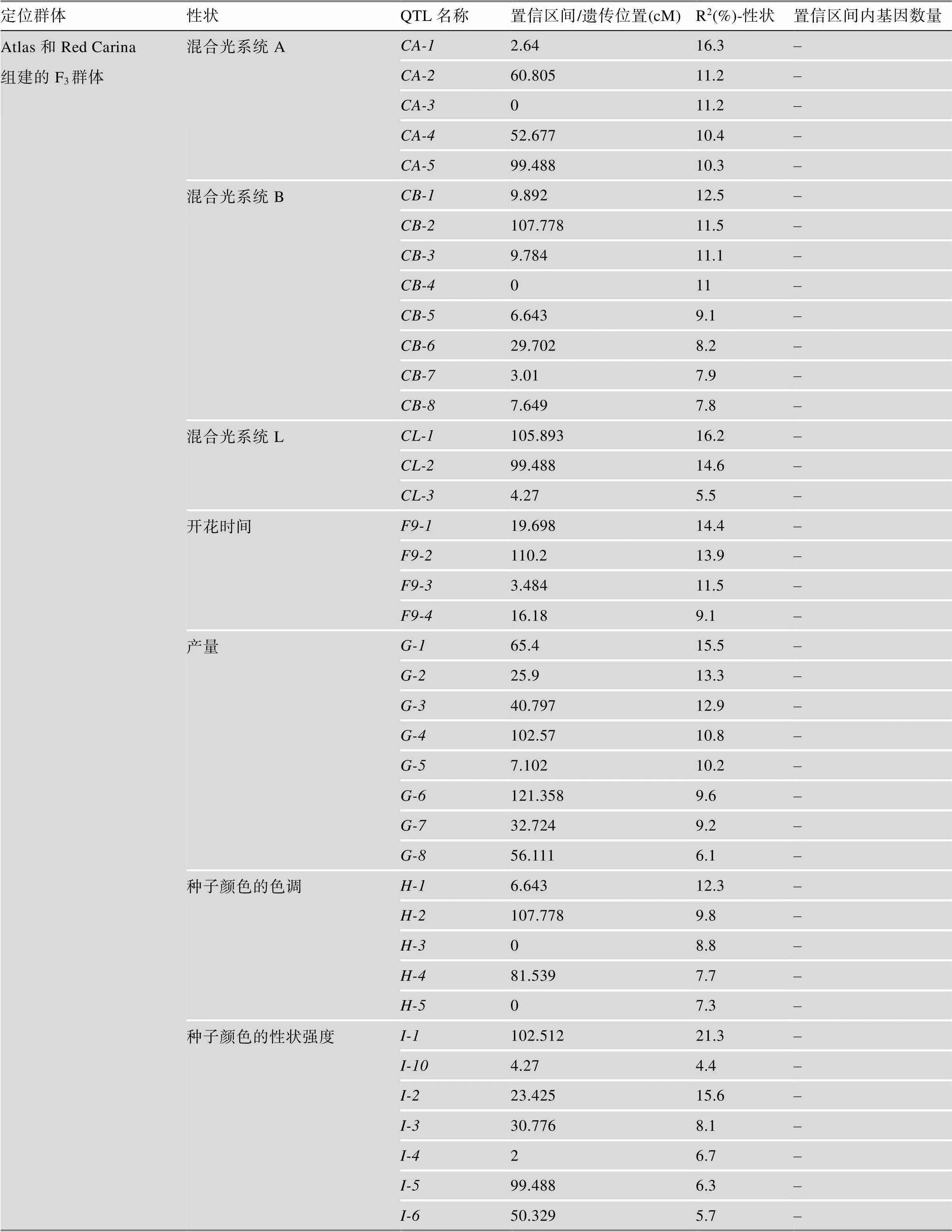
续表
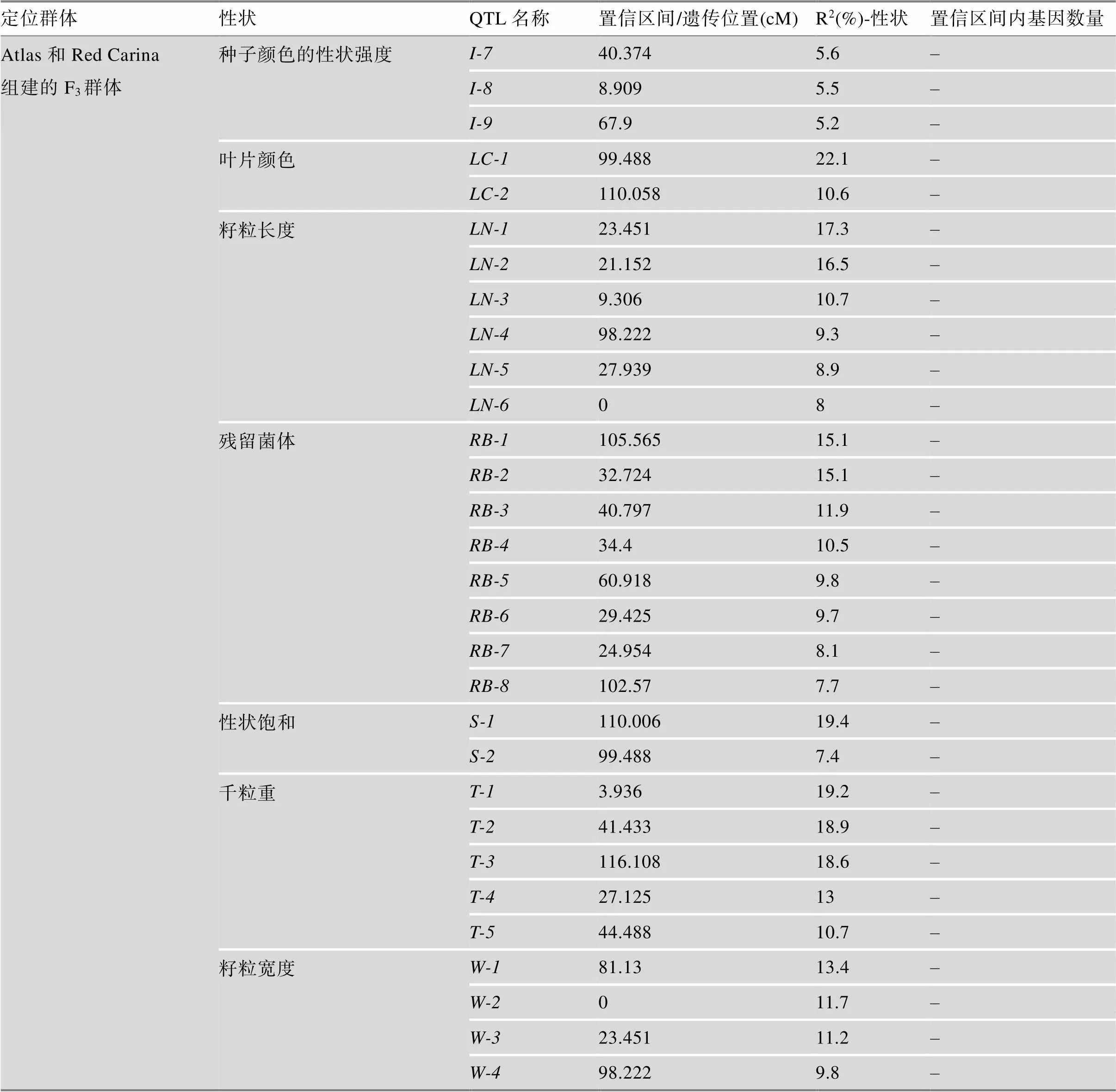
续表
表中数据整合自参考文献[30,32],DTF、PH、TKW、DTM、PL、PD、MS、SW和SN分别表示开花日数、株高、千粒重、成熟日数、穗长、穗密度、霉病易感性、单株粒重和每株种子数。
4 藜麦遗传改良重要基因研究进展
目前藜麦遗传转化体系尚未建立,各性状相关基因的研究仍局限于物种的同源比对及基因的表达分析,功能基因的挖掘和鉴定在藜麦中研究甚少。本文主要针对藜麦生长发育、农艺性状、品质、非生物抗性及生物抗性这5个方面涉及的主要性状以及各性状相关基因的研究进展进行阐述,根据其他作物中已定位基因预测的藜麦各性状相关基因详见图1和表4。
4.1 藜麦开花相关基因的研究进展
藜麦的栽培范围已经扩展到安第斯山脉以外的世界各地,确定藜麦对不同白昼长度响应的遗传因子对更好地开发适应新环境的新品种有着决定性意义。2017年,Jarvis等[6]在藜麦基因组中发现了拟南芥()调节开花时间(flowering time,FT)基因的两个同源基因和,两个基因分别有3个和2个同源基因,其中的3个同源基因以串联重复的形式存在。进一步的功能研究需要确定藜麦是否起促进开花作用,而是否在春化前起抑制开花作用[33,34]。另外,根据最近发表的藜麦全基因组序列,通过序列相似性挖掘藜麦中开花相关基因,从而促进对藜麦开花调控的理解。
4.2 藜麦农艺性状相关基因的研究进展
4.2.1 株高
株高与作物的株型、生物量、产量等重要性状密切相关。适当降低株高可以增强作物的抗倒伏能力,提高收获指数,从而实现作物的增产与稳产。20世纪以来,小麦中()和()[35]以及玉米中和[36]的广泛使用,使全世界小麦和玉米产量大幅度提升,极大地保障了全球粮食安全。在南美洲,藜麦植株可达3 m,倒伏是影响藜麦产量的潜在的问题,有报道显示株高与种子产量呈负相关[37]。因此,克隆影响株高的基因是提高藜麦产量的目标之一。在藜麦基因组中存在小麦/的两个同源基因(和),这些基因也是拟南芥(编码一个参与赤霉素信号转导的转录因子)的同源基因,是有望改良藜麦株高的重要基因[38]。
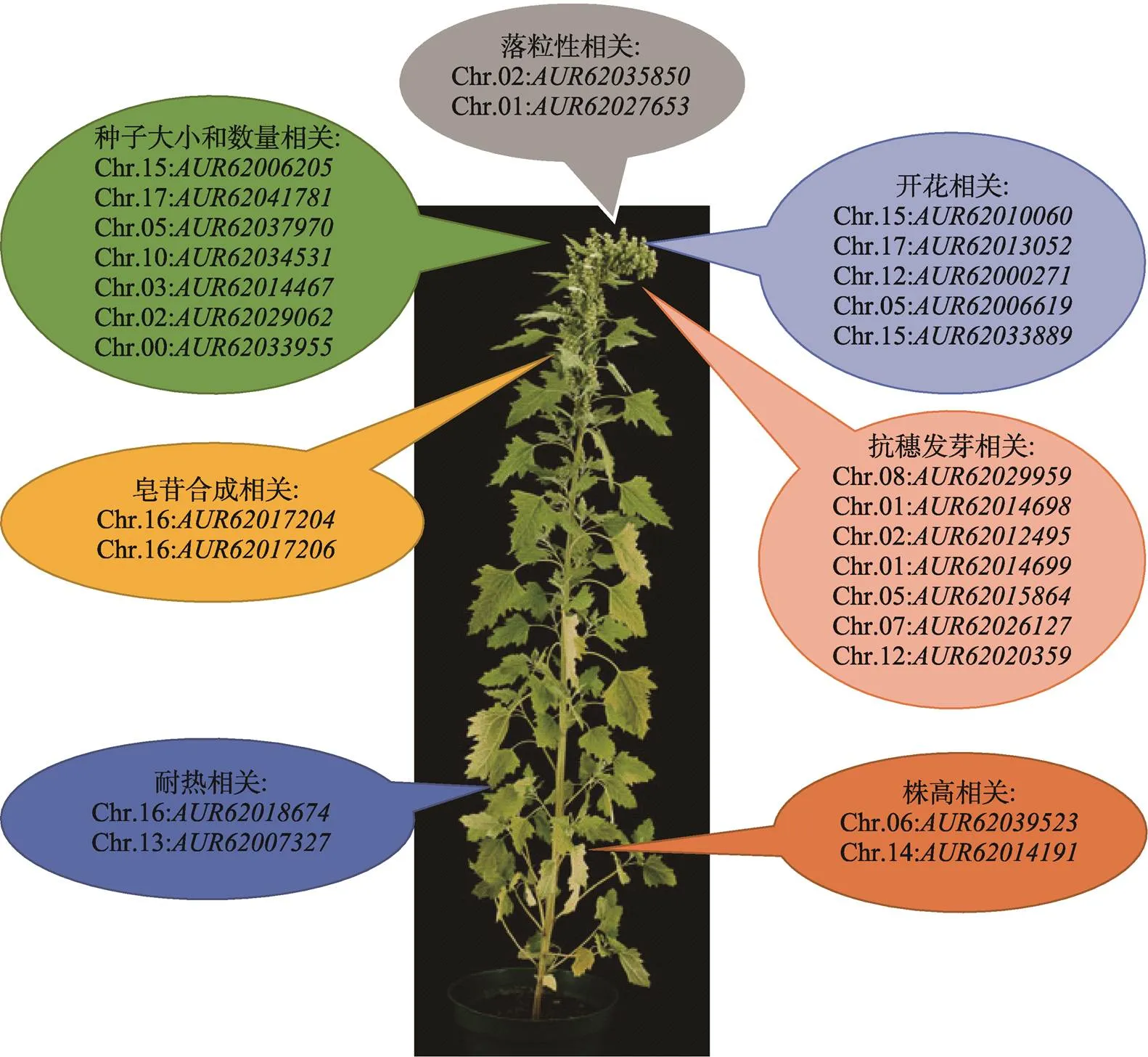
图1 藜麦重要性状相关基因的预测

表4 藜麦重要性状相关基因的预测
NA:未找到相关数据。
4.2.2 粒重
植物种子大小作为重要的产量构成要素,长期以来一直作为作物育种改良的主要目标之一。研究表明,水稻(L.)中有控制种子大小的基因,包括(,编码一个环型E3泛素连接酶)、(,编码籽粒早期灌浆过程中碳分配所需的细胞壁转化酶)[39]。是导致野生稻()籽粒较小的基因,基因的累积突变导致驯化水稻品种的籽粒变大。是拟南芥的同源基因,是细胞分裂的负调控因子,功能缺失突变体的细胞数量增加,导致小穗壳变宽。通过基因的同源比对,仅在藜麦中鉴定到的一个同源基因,存在两个同源基因和。因此,研究藜麦同源基因的功能缺失突变将是增加藜麦种子大小的一个切入点。
在相同的遗传背景下,将增加粒数和种子大小的基因组合在一起,可为作物的定向改良提供策略。在水稻中,()和()功能缺失突变的组合产生杂交优良水稻的重穗表型[40]。在拟南芥中,通过缺失水稻两个同源基因和可以模拟水稻突变表型[41]。基因同源比对发现,藜麦中存在的两个相近的同源基因(和)和的另两个同源基因(和),这可能是提高藜麦种子产量潜在的目标基因。
4.2.3 落粒性
落粒性是野生植物在种子成熟时释放并繁衍后代的一种能力,这种能力对植物在野外生存至关重要,但会给农业生产系统造成巨大损失。一般来说,驯化作物的种子传播机制失活,造成脱落区周围细胞壁较厚,导致种子或果实无法从母组织分离[42]。目前,已经在许多物种中报道了控制种子落粒性的关键基因。在水稻中,、和协同作用控制分离区发育,其中是主要的转录因子,正向调控的活性,影响上述两种转录因子的表达[43]。在拟南芥中,冗余的MADS-box转录因子()和负责裂缝区分化和种子传播[44]。在藜麦中存在的两个同源基因(和)。尽管其他作物中已经报道了控制落粒性相关的基因,但是藜麦中还未有相关的报道,这也是藜麦未来研究的一个重要方向。
4.3 藜麦品质相关基因的研究进展
4.3.1 皂苷
藜麦种子通常含有皂苷(三萜苷的混合物),皂苷对植物生长具有一定的驱虫效应,但其味苦且对人体有一定的毒害。目前生产无皂苷藜麦种子的策略是通过湿法或干法来去除它们,这两种方法分别需要水浸泡种子或专门的机械处理,耗时且成本较高。利用藜麦种质资源中天然皂苷含量较低品种的遗传多样性,开发无皂苷藜麦品种将是一种有前景的替代方法。然而,实现这一策略的前提是克隆藜麦中调控皂苷合成的基因。基于这一目标,Fiallos-Jurado等[45]结合生理学和分子生物学的方法,从厄瓜多尔甜藜和苦藜基因型中鉴定参与藜麦皂苷生物合成的新基因。该研究首先证实了茉莉酸甲酯(MeJA)处理会诱导藜麦叶中皂苷的合成,然后利用两个公开的藜麦RNA-seq数据集进行了重新转录组组装,以确定22个已知在拟南芥中稳定表达的藜麦同源序列,并作为藜麦qPCR分析及藜麦皂苷生物合成候选基因筛选的参考。
2017年,Jarvis等[6]利用Kurmi(甜藜)×0654(苦藜)和Atlas(甜藜)×Carina Red(苦藜)分别构建两个分离群体,通过连锁图谱定位和集群分离分析(bulked segregant analysis,BSA),在CqB16染色体上鉴定到一个种子皂苷存在/缺失相关的位点。在该区域内,两个转录因子基因和根据参考基因组注释为basic helix-loop-helix(bHLH),与三萜皂苷生物合成激活调控因子()基因相似,该基因在皂苷生物合成中发挥作用。利用RNA-seq测定并分析了这些基因在藜麦根、花和未成熟种子组织中的表达,发现(、)在藜麦根中有组织特异性表达,而()在苦味品种的种子中表达更高。此外的序列包含多个独立的突变,且与甜味表型共分离,进一步确定了是调节藜麦种子中皂苷存在和缺失的关键候选基因。
4.3.2 种子贮藏蛋白
藜麦的主要品质之一在于其种子含有丰富的蛋白质且氨基酸含量均衡,因此挖掘藜麦种子高品质的关键基因能为藜麦育种提供重要线索。2008年,Balzotti等[46]从藜麦发育种子构建的cDNA文库中筛选分离出两个11S基因的cDNA序列,测序结果表明,这两个基因分别属于藜麦基因组的同源位点11SA和11SB,是一个编码11S种子贮藏蛋白的两个变体,与的11S球蛋白同源性达74%。通过对藜麦11S氨基酸序列的分析,发现与其他物种的氨基酸序列具有保守性,并鉴定出一个定位于内质网(ER)的包含25个氨基酸残基的信号肽。基因表达分析表明,不同地理来源和成熟速率的9种藜麦基因型种子成熟后期11S mRNA表达量较高。Balzotti等[47]论证了11S mRNA的表达水平与种子中11S蛋白的积累和成熟速率相关,这与其他植物种子发育过程中贮藏蛋白积累的模式一致。编码藜麦第二大种子贮藏2S蛋白基因的克隆、测序和表达特征有待进一步研究,该基因与11S种子蛋白结合产生了藜麦种子蛋白质的独特数量和组成。
4.4 藜麦非生物抗性相关基因的研究进展
藜麦种植在应对气候变化和人口增长方面对全球粮食安全的重要性日益突出,研究关键增强耐受性的基因,阐明藜麦对非生物胁迫耐受性的遗传基础有助于制定具体、有效和快速的育种计划。最近发布的高质量藜麦参考基因组为非生物逆境抗性基因的挖掘提供了重要的工具。有关藜麦非生物抗性研究虽然在遗传水平上的研究相对较少,但现有的研究对未来藜麦的遗传改良仍具有重要指导意义。
4.4.1 耐盐
植物的耐盐性是一种重要的非生物抗性。藜麦是一种兼性盐生作物,其耐盐性可达150~300 mmol/L NaCl,耐盐程度大大高于大麦(L.)、小麦(L.)和玉米(L.)[48]。利用EST序列资源克隆了藜麦的两个盐过度敏感(salt overly sensitive 1,)基因和,两个基因均编码一个质膜Na+/H+逆向转运体。序列分析表明,和基因各有23个外显子,分别包含3477 bp和3486 bp的编码序列,这些序列与其他物种的同源物有高度的相似性,包含两个保守的结构域,一个阳离子-反向转运体结构域(Nhap)和一个环核苷酸结合结构域。在盐水条件下(450 mmol/L),在根和叶组织中均有相对表达量,叶中的相对表达量始终是根的3~4倍,而且叶片对盐胁迫的反应比根更强烈,这表明胁迫下基因在根中具有结构性表达,而在叶中具有诱导表达,因此对盐渍环境下藜麦的萌发和生长中起重要作用[49]。
Schmockel等[50]将RNA测序分析与比较基因组学和蛋白质拓扑预测方法相结合,以识别藜属植物品种PI614886基因组参考序列中涉及耐盐的候选基因。作者通过盐胁迫下的差异表达基因共鉴定出219个候选基因,这些候选基因相对于其他苋菜科物种来说具有较高的特异性,均含有一个及以上预测的跨膜结构域。比较了21份藜属植物(14份、5份和2份)的219个候选基因中的单核苷酸多态性和拷贝数变异(copy number variation,CNV)及其对盐度的响应,发现了15个可能影响藜属植物耐盐性差异的基因,这些候选基因是未来提高藜麦耐盐性的研究目标。
藜麦可以在高盐环境中生长,得益于其植株表面独特的表皮组织(epidermal bladder cells,EBCs)。为了探究盐如何进入EBCs的分子机制,Bohm等[51]在存在土壤盐度(处理)和不存在土壤盐度(对照)的情况下,收集了5020株藜麦植物的EBCs并进行转录组测序,并对囊泡表达转运蛋白进行了功能分析。确定了在藜麦EBCs中差异表达的膜转运蛋白编码基因,证实了为使盐进入并在其液泡中积累,Na+和Cl–需要穿过细胞膜到达囊泡细胞质,文章还绘制了极性盐转运系统的工作模型。Imamura等[52]鉴定了盐生藜属植物中参与EBC形成的基因,并揭示了新的EBC功能。实验证实编码一个WD40蛋白,该蛋白定位于细胞核和叶绿体。此外,系统发育和转基因植物分析显示,REBC蛋白不同于TTG1,参与毛状体的形成,这些研究结果为了解EBC的形成机制提供了依据,同时该结果有助于阐明含EBC盐生植物的抗逆性机制。
4.4.2 抗旱
藜麦能够适应安第斯山脉及其他地区不断变化的环境,在半干旱甚至沙漠条件下生长并收获种子,因此了解藜麦对干旱胁迫的响应机制及其分子基础可以为培育抗旱广适的藜麦新品种奠定基础。Liu等[53]在藜麦基因组中鉴定出16个编码70 kDa热休克蛋白(Hsp70s)的基因,该基因是一组保守的伴侣蛋白,长度为412 aa~891 aa,在多种植物中已被证实在干旱胁迫耐受中发挥作用。Morales等[9]的一项研究检测了智利3种藜麦基因类型在干旱条件下的转录反应。他们根据其相对含水量,电解质泄漏和光系统II的最大效率,确定基因型R49(一个salares品种)是最耐旱的。值得注意的是,sHSP家族基因的表达高度上调。的表达增加了200倍,表明它是藜麦对干旱胁迫的反应的一部分。同时该研究还报道了水稻中被证明对ABA敏感的基因也表现为高度上调表达。(通过ABA依赖和不依赖的途径在调节植物对胁迫的适应中发挥关键作用)上调表达,表明它可能需要协调支撑非生物抗逆性的调控网络[54]。
4.4.3 耐热
炎热的气候和沙漠会给藜麦的种植带来很大的挑战,导致花粉活力降低,籽粒不能正常结实。由于北美大部分温带地区的夏季温度都超过了藜麦的耐受阈值,开发耐热品种有利于扩大藜麦的生产面积,并提高热浪破坏性地区的收获安全性。HSFA1s是热休克因子(heat shock factor,HSF)蛋白家族成员之一,已成为影响植物热休克反应的主要转录因子,参与了植物获得热耐受性的关键转录级联调节[55]。通过基因比对发现藜麦基因组中存在的两个相近同源基因(和)。2019年,Hinojosa等[56]在玻璃温室对112种藜麦基因型的筛选及其随后的田间评价表明,它们在耐热性方面存在显著的遗传变异。因此,对这些材料进行全基因组关联研究(genome-wide association studies,GWAS)分析和/或基因组测序可能揭示藜麦不同耐热性的分子基础。
4.4.4 耐冷
藜麦在海拔超过4000 m的安第斯山脉种植了数千年,对霜冻和寒冷胁迫具有相对较高的耐受性,但该耐受性取决于品种、发育阶段和冷胁迫程度。了解藜麦在发育后期如何以及为什么失去对低温胁迫的耐受性,可能在气候变化中保护藜麦农业具有重要意义。Morales等[9]指出,CqCAP160(冷适应蛋白)在干旱条件下增加,表明CqCAP160对胁迫的反应更为普遍。目前还没有关于CqCAP160在温度胁迫耐受中的作用的研究。尽管对藜麦抗低温的生理和机理进行了研究,但对遗传水平知之甚少,例如糖转化酶、过冷和细胞壁酶的遗传控制。随着近年来高质量藜麦基因组的公布,可以通过对藜麦和拟南芥进行比较基因组学分析,识别感兴趣的耐冷位点。
4.4.5 抗除草剂
杂草控制对藜麦生产有重大影响,是限制藜麦扩张的主要因素之一,提高产量的第一步是实现有效的杂草控制[57]。鉴于此,Mestanza等[58]对智利沿海藜麦品种(Regalona-Baer)的乙酰羟基酸合成酶(acetohydroxyacid synthase,)基因家族进行了鉴定和分析,为开展抗除草剂育种提供基础信息。基因是编码生物合成途径中产生缬氨酸、亮氨酸和异亮氨酸必需氨基酸支链的第一种酶。由于该基因可能在多倍体物种中存在多个拷贝,因此研究该基因家族对于通常只有一个基因拷贝的杂草二倍体物种中开发针对该酶的除草剂抗性具有特殊的意义。Mestanza等[58]对基因进行了克隆、测序和印迹杂交,发现在藜麦四倍体基因组中有6个拷贝。通过与NR数据库中mRNA序列的比较发现,和是最保守的基因拷贝,它们可能分别从不同的二倍体亲本遗传而来,并且可能是多倍体事件发生后产生的重复拷贝的起点。用最大简约系统发育学进一步分析表明,和是功能性的,而的表达情况还不清楚,因此还需要进行进一步的基因表达分析,以确定其他变异是否以组织特异性的方式表达。
4.4.6 抗穗发芽
在夏季多雨的国家(例如北欧国家)种植藜麦时,收获前发芽(pre-harvest sprouting)是一个重要的挑战,因为穗发芽会严重影响籽粒的产量和品质[59]。调节谷物休眠期长短是控制收获前穗发芽的有效策略,未来的育种需要设计更适应区域气候的品种。拟南芥()编码一种磷脂酰乙醇胺结合蛋白,该蛋白调节拟南芥种子萌发[60]。通过系统发育分析,López-Marqués等[61]在藜麦中鉴定出的一个同源基因(),同源度高达73.41%,此外还有3个基因 (、和)与有一定的同源性,具体功能需要进一步确定。研究表明,(mitogen-activated protein kinase kinase 3)分别是大麦和小麦籽粒主要休眠数量性状位点()和的关键基因[62]。序列同源比对发现,藜麦基因组编码3个与相近的同源基因(、和),这些基因都对减少穗发芽发生有一定作用[57]。
4.5 藜麦抗病虫基因的研究进展
除了非生物胁迫,生物胁迫也会影响植物的生长和生存。因此,研究藜麦对病原菌和害虫的反应,鉴定抗性或低易感基因型,对藜麦育种具有重要意义。
4.5.1 抗病
霜霉病由引起,是对藜麦最具破坏性的病原体,会导致产量大幅下降,但抗性机制尚不清楚[63]。一些研究试图通过田间试验筛选不同国家的多个品种对霜霉病的抗性,结果发现这些品种的抗病性存在广泛的变异同时也鉴定到许多抗病基因型,但是由于抗病基因型起源地包括智利沿海低地和秘鲁,所以并没有发现抗病性和地理起源之间存在明显的相关性[64,65]。Ochoa等[66]从厄瓜多尔鉴定出60份藜麦种质资源与24株霜霉病分离株。该研究确定了霜霉病的不同毒力群和藜麦的抗性因子,能够对抗病的类型和程度进行分类。此外,该研究鉴定了对某些分离株具有抗性的种质,发现没有对所有分离株都具有抗性的种质。这项研究表明,藜麦种质和病原体都是高度可变的,抗病种质不能对病原体的所有菌株都产生抗性。因此未来的研究需要对藜麦的防御机制进行深入研究,以确定改良品种的潜在遗传目标,并与植物抗性的遗传评估相辅相成。
4.5.2 抗虫
昆虫对藜麦的危害极大,尤其是当它们以花的结构或种子为食时,会降低种子的产量和质量。由于昆虫和植物之间存在着复杂的形态、生化和生理相互作用,抗虫育种必须考虑害虫和宿主的遗传学关系。植物对虫害的抗性主要分为3种:第一是通过抑制摄食或产卵(非偏好);第二是对昆虫的正常生长或生存产生不利影响(抗生素);第三是有昆虫时植物仍具备抗虫能力来获得生存(耐受性)。目前,在藜麦的生产地已报道了许多害虫,包括甜菜夜蛾、甲虫和鼠李蚜、船瘿蚜虫、甜菜根蚜虫等[67]。尽管已经发现了许多影响藜麦生长和产量的害虫,但是关于藜麦的害虫防治及抗虫藜麦品种的挖掘仍然处于起始阶段,未来需要人们更加深入地鉴定藜麦品种中的抗虫基因,积极改良培育新的抗虫藜麦品种,有效提高藜麦的产量和品质。
5 结语与展望
5.1 藜麦生产研究存在的问题
藜麦具有很高的生物学价值,富含矿物质和维生素,含有高质量的蛋白质,包括人体营养必需的所有氨基酸,被认定为超级食物[68]。由于藜麦可以生长在极端气候和土壤下,特别是霜冻、干旱和盐碱地区,藜麦也被认为是一种超级作物[69]。虽然藜麦有诸多优点,但藜麦作为超级食物和超级作物在遗传研究、育种方法、种质资源创新、不同用途品种培育与加工技术方面的研究均处于初级阶段,仍有一些关键的问题需解决:(1)与谷类作物相比,藜麦不耐除草剂,不抗倒伏,易穗发芽,产量较低;(2)藜麦的食用受到种皮中积累的皂苷的限制,皂苷是一种抵御病虫害的防御机制,必须在食用前去除,工序较为繁琐;(3)大多数甜藜麦品种对霉病及虫害极为敏感,造成较大的产量损失;(4)藜麦不耐热,严重限制了藜麦的种植范围及产量的提高;(5)通过基因工程培育转基因藜麦仍然极具挑战性。
5.2 未来藜麦遗传改良和育种的重要方向
5.2.1 加强种质资源筛选收集,积累丰富的变异资源
种质资源是育种的重要基础,能够为品种改良带来丰富的变异资源,种质资源的多少直接决定育种质量的高低。藜麦种质资源收集、创新、保存和鉴定工作相对落后,优异种质匮乏,缺少适应性强、耐热、抗倒伏、耐穗发芽、高产等优质的种质资源,限制了藜麦产业的健康可持续发展。人类在藜麦驯化中常追求高产、高品质和成熟一致等目标,导致藜麦遗传多样性的降低或丢失,造成了现有遗传改良的瓶颈问题日益突出[70]。因此核心种质资源收集及创新是藜麦产业链的第一个环节。作为藜麦原产国,南美洲国家保存了大量的藜麦种质资源材料,其中以玻利维亚和秘鲁最多,均超过了5000份。引进和收集不同国家、不同生态区不同类型的藜麦种质资源是藜麦育种的重要途径[71]。此外由于藜麦是异源多倍体,收集二倍体栽培种和野生近缘和等种质起源,有助于恢复藜麦种质资源的遗传多样性,有效实现藜麦的遗传改良。
5.2.2 加强藜麦精准人工杂交育种创新技术研究
针对藜麦花序繁复、异花授粉率高、自身变异大、杂交父母本选择有限等无法进行精准杂交的技术问题以及藜麦杂交育种技术滞后的实际问题,应开展藜麦精准人工杂交育种创新技术研究,筛选优质父母本种质材料,建立对母本藜麦材料的修穗去雄、人工授精、去杂保种等操作技术规范,精准获得目标性状杂交后代;创制雄性不育系特色藜麦种质材料,利用雄性不育系配制杂交种,降低杂交种子生产成本,提高杂种质量,扩大杂种优势的利用范围,建立最优化制种途径,培育耐热、抗倒伏、高产优质藜麦新品种。
5.2.3 藜麦分子育种研究有待突破
新的育种技术的出现,尤其是CRISPR/Cas系统,可以同时精确编辑多个基因或等位基因,为藜麦的靶向分子育种提供了良好的平台[72]。然而,由于藜麦是含有A和B基因组的异源四倍体,目前还没有成功建立遗传转化体系,使分子育种进程受阻。因此,未来的研究通过加强不同转化手段的基础研究,可借鉴麦类作物进行幼胚转化体系的方法或直接用农杆菌浸花等手段尝试遗传转化,旨在通过转基因手段进行重要性状基因或QTL功能的确定及育种应用的研究。
高质量基因组数据的公布,为深入研究藜麦的特殊营养价值提供了基础,并为定向培育藜麦新品种开辟了可能。藜麦的产量(种子大小和数量的增加)、品质(高蛋白、低皂苷)、开花时间、对病虫害的抗性和对非生物胁迫(盐、旱、热、冷、除草剂等)的适应等因素均是未来研究需要改变的重要性状。因此,未来的研究应大力应用单倍体育种技术、EMS诱变技术、高通量测序和基因编辑技术,旨在加强常规和分子育种相结合的方法,加速藜麦的“5G”育种,即通过基因组研究(genome)、种质资源鉴定(germplasm)、基因功能鉴定(gene function)、基因组育种(genomic breeding)和基因编辑(gene editing),显著加速藜麦的高产、高品质、高抗逆、低消耗育种,构建藜麦高产、高效、高质、高值农业模式。
[1] Organization FA. Genebank standards for plant genetic resources for food and agriculture., 2013, 11(1): 1–16.
[2] Yan F, Li QQ, Dong Y, Ji SD. Industry status and development countermeasures of, 2021, (9): 98–100.
闫锋, 李清泉, 董扬, 季生栋. 藜麦产业现状及发展对策. 黑龙江农业科学, 2021, (9): 98–100.
[3] Ren GX, Yang XS, Me Y. Current situation of Chinese quinoa industry., 2015, (5): 1–5.
任贵兴, 杨修仕, 么杨. 中国藜麦产业现状. 作物杂志, 2015, (5): 1–5.
[4] Kolano B, McCann J, Orzechowska M, Siwinska D, Temsch E, Weiss-Schneeweiss H. Molecular and cytogenetic evidence for an allotetraploid origin ofand().,2016, 100: 109–123.
[5] Yasui Y, Hirakawa H, Oikawa T, Toyoshima M, Matsuzaki C, Ueno M, Mizuno N, Nagatoshi Y, Imamura T, Miyago M, Tanaka K, Mise K, Tanaka T, Mizukoshi H, Mori M, Fujita Y. Draft genome sequence of an inbred line of, an allotetraploid crop with great environmental adaptability and outstanding nutritional properties., 2016, 23(6): 535–546.
[6] Jarvis DE, Ho YS, Lightfoot DJ, Schmöckel SM, Li B, Borm TJA, Ohyanagi H, Mineta K, Michell CT, Saber N, Kharbatia NM, Rupper RR, Sharp AR, Dally N, Boughton BA, Woo YH, Gao G, Schijlen EGWM, Guo XJ, Momin AA, Negrão S, Al-Babili S, Gehring C, Roessner U, Jung C, Murphy K, Arold ST, Gojobori T, van der Linden CG, van Loo EN, Jellen EN, Maughan PJ, Tester M. The genome of., 2017, 542(7641): 307–312.
[7] Wang KY, Li L, Li SK, Sun HH, Zhao MZ, Zhang MP, Wang Y. Characterization of the complete chloroplast genome ofWilld., 2017, 2(2): 812–813.
[8] Stevens MR, Coleman CE, Parkinson SE, Maughan PJ, Zhang HB, Balzotti MR, Kooyman DL, Arumuganathan K, Bonifacio A, Fairbanks DJ, Jellen EN, Stevens JJ. Construction of a quinoa (Willd.) BAC library and its use in identifying genes encoding seed storage proteins., 2006, 112(8): 1593– 1600.
[9] Morales A, Zurita-Silva A, Maldonado J, Silva H. Transcriptional responses of Chilean quinoa (Willd.) under water deficit conditions uncovers ABA-independent expression patterns., 2017, 8: 216.
[10] Liu Q, Zhang GY, Chen SY. Structure and regulatory function of plant transcription factors., 2001, 46: 271–278.
[11] Alshareef NO, Rey E, Khoury H, Tester M, Schmöckel SM. Genome wide identification of NAC transcription factors and their role in abiotic stress tolerance in., 2019, Doi: 10.1101/693093.
[12] Zhang DL. Identification and analysis of quinoa MADS-box gene family and study on agrobacterium- mediated root transformation[Dissertation]. Yantai University, 2021.
张东亮. 藜麦MADS-box基因家族的鉴定与分析及发根农杆菌介导的根转化研究[学位论文]. 烟台大学, 2021.
[13] Yue H, Chang X, Zhi YQ, Wang L, Xing GW, Song WN, Nie XJ. Evolution and identification of the WRKY gene family in quinoa ()., 2019, 10(2): 131.
[14] Li KY, Fan Y, Zhou GY, Liu XJ, Chen SS, Chang XC, Wu WQ, Duan LL, Yao MX, Wang R, Wang ZL, Yang MF, Ding YQ, Ren MJ, Fan Y, Zhang LY. Genome-wide identification, phylogenetic analysis, and expression profiles of trihelix transcription factor family genes in quinoa (Willd.) under abiotic stress conditions., 2022, 23(1): 499.
[15] Zhang YM, Zhu LL, Chen ZG. Identification and expression analysis ofgene family in quinoa under salt stress., 2022, 1–10.
张业猛, 朱丽丽, 陈志国. 藜麦NHX基因家族鉴定及盐胁迫下表达分析. 生物技术通报, 2022, 1–10.
[16] Xiao YL. Identification analysis and functional vertification ofgene family in[Dissertation]. Shandong Normal University, 2021.
肖玉林. 藜麦TCP基因家族鉴定分析与功能验证[学位论文]. 山东师范大学, 2021.
[17] Cao HQ. Isolation and molecular characterization of quinoafamily genes[Dissertation]. Shanxi University, 2021.
曹华麒. 藜麦SPL家族基因的分离和分子特性分析[学位论文]. 山西大学, 2021.
[18] Tashi G, Zhan HS, Xing GW, Chang X, Zhang H, Nie XJ, Ji WQ. Genome-wide identification and expression analysis of heat shock transcription factor family inWilld., 2018, 8(7): 103.
[19] Shi PB, He B, Fei YY, Wang J, Wang WY, Wei FY, Lv YD, Gu MF. Identification and expression analysis oftranscription factor family of., 2019, 45(12): 1841–1850.
时丕彪, 何冰, 费月跃, 王军, 王伟义, 魏福友, 吕远大, 顾闽峰. 藜麦GRF转录因子家族的鉴定及表达分析. 作物学报, 2019, 45(12): 1841–1850.
[20] Zhu MX, Yang YS, Yang XL, Zhang F, Yang LY, Wang CY. Identification and expression analysis oftranscription factor family of., 2020, 43(4): 43–49.
朱满喜, 杨雅舒, 杨小兰, 张芳, 杨利艳, 王创云. 藜麦WOX转录因子家族的鉴定及表达分析. 湖南师范大学自然科学学报, 2020, 43(4): 43–49.
[21] Zhang DL, Wu XL, Tian XQ, Chu J, Guo SL, Chen SH. Identification and expression ofgene family in., 2021, 34(4): 400–405.
张东亮, 吴筱林, 田晓芹, 褚晶, 郭善利, 陈世华. 藜麦基因家族的鉴定及表达. 烟台大学学报(自然科学与工程版), 2021, 34(4): 400–405.
[22] Shi XD, Sun MH, Wu Q, Tian YS, Xu Y. Identification and expression analysis of fatty acid export gene family in, 2020, 39(12): 5652–5659.
时小东, 孙梦涵, 吴琪, 田银帅, 徐莺. 藜麦脂肪酸转运基因家族FAX的鉴定与表达分析. 基因组学与应用生物学, 2020, 39(12): 5652–5659.
[23] Zhu MX, Zhang YR, Yang YS, Yang XL, Wang CY, Deng Y, Zhao L, Zhang LG, Qin LX, Yang LY. Identification and expression analysis oftranscription factor family ofWilld., 2021, 36(4): 37–46.
朱满喜, 张玉荣, 杨雅舒, 杨小兰, 王创云, 邓妍, 赵丽, 张丽光, 秦丽霞, 杨利艳. 藜麦NLP转录因子家族的鉴定及表达分析. 华北农学报, 2021, 36(4): 37–46.
[24] Maughan PJ, Smith SM, Rojas-Beltrán JA, Elzinga D, Raney JA, Jellen EN, Bonifacio A, Udall JA, Fairbanks DJ. Single nucleotide polymorphism identification, characterization, and linkage mapping in quinoa., 2012, 5(3): 114–125.
[25] Fairbanks DJ, Waldrigues A, Ruas CF, Maughan PJ, Robinson LR, Andersen WR, Riede CR, Pauley CS, Caetano LG, Arantes OMN, Fungaro MHP, Vidotto MC, Jankevicius SE. Efficient characterization of biological diversity using field DNA extraction and random amplified polymorphic DNA markers., 1993, 16(1): 11–22.
[26] Maughan PJ, Bonifacio A, Jellen EN, Stevens MR, Coleman CE, Ricks M, Mason SL, Jarvis DE, Gardunia BW, Fairbanks DJ. A genetic linkage map of quinoa () based on AFLP, RAPD, and SSR markers., 2004, 109(6): 1188–1195.
[27] Mason SL, Stevens MR, Jellen EN, Bonifacio A, Fairbanks DJ, Coleman CE, Mccarty RR, Rasmussen AG, Maughan PJ. Development and use of microsatellite markers for germplasm characterization in quinoa (Willd.)., 2005, 45(4): 1618–1630.
[28] Zhang TF, Gu MF, Liu YH, Lv YD, Zhou L, Lu HY, Liang SQ, Bao HB, Zhao H. Development of novel InDel markers and genetic diversity inthrough whole-genome re-sequencing., 2017, 18(1): 685.
[29] Jarvis DE, Kopp OR, Jellen EN, Mallory MA, Pattee J, Bonifacio A, Coleman CE, Stevens MR, Fairbanks DJ, Maughan PJ. Simple sequence repeat marker development and genetic mapping in quinoa (Willd.)., 2008, 87(1): 39–51.
[30] Cervantes DP, Van Loo E.QTL Mapping for agromorphological traits in quinoa (Willd.). Wageningen University, 2017.
[31] Patiranage DSR, Rey E, Emrani N, Wellman G, Schmid K, Schmöckel SM, Tester M, Jung C. Genome-wide association study in quinoa reveals selection pattern typical for crops with a short breeding history., 2020, 11: e66873.
[32] Maldonado-Taipe N, Barbier F, Schmid K, Jung C, Emrani N. High-density mapping of quantitative trait loci controlling agronomically important traits in quinoa (Willd.)., 2022, 13: 916067.
[33] Pin PA, Nilsson O. The multifaceted roles ofin plant development., 2012, 35(10): 1742–1755.
[34] Golicz AA, Steinfort U, Arya H, Singh MB, Bhalla PL. Analysis of the quinoa genome reveals conservation and divergence of the flowering pathways., 2020, 20(2): 245–258.
[35] Peng J, Richards DE, Hartley NM, Murphy GP, Devos KM, Flintham JE, Beales J, Fish LJ, Worland AJ, Pelica F, Sudhakar D, Christou P, Snape JW, Gale MD, Harberd NP. 'Green revolution' genes encode mutant gibberellin response modulators., 1999, 400(6741): 256–261.
[36] Winkler RG, Freeling M. Physiological genetics of the dominant gibberellin-nonresponsive maize dwarfs,and., 1994, 193: 341–348.
[37] Maliro MFA, Guwela VF, Nyaika J, Murphy KM. Preliminary studies of the performance of quinoa (Willd.) genotypes under irrigated and rainfed conditions of central Malawi., 2017, 8: 227.
[38] Silverstone AL, Ciampaglio CN, Sun T. The Arabidopsisgene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway., 1998, 10(2): 155–169.
[39] Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase., 2007, 39(5): 623–630.
[40] Wang SG, Ma BT, Gao Q, Jiang GJ, Zhou L, Tu B, Qin P, Tan XQ, Liu PX, Kang YH, Wang YP, Chen WL, Liang CZ, Li SG. Dissecting the genetic basis of heavy panicle hybrid rice uncoveredandas key genes., 2018, 131(6): 1391–1403.
[41] Bartrina I, Otto E, Strnad M, Werner T, Schmülling T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in., 2011, 23(1): 69–80.
[42] Ballester P, Ferrándiz C. Shattering fruits: variations on a dehiscent theme., 2017, 35: 68–75.
[43] Hofmann NR., a new player in seed shattering of rice., 2012, 24(3): 839.
[44] Liljegren SJ, Ditta GS, Eshed Y, Savidge B, Bowman JL, Yanofsky MF. Shatterproof MADS-box genes control seed dispersal in., 2000, 404(6779): 766–770.
[45] Fiallos-Jurado J, Pollier J, Moses T, Arendt P, Barriga- Medina N, Morillo E, Arahana V, de Lourdes Torres M, Goossens A, Leon-Reyes A. Saponin determination, expression analysis and functional characterization of saponin biosynthetic genes inleaves., 2016, 250: 188–197.
[46] Balzotti MRB, Thornton JN, Maughan PJ, Mcclellan DA, Stevens MR, Jellen EN, Fairbanks DJ, Coleman CE. Expression and evolutionary relationships of the11S seed storage protein gene., 2008, 169(2): 281–291.
[47] Nakamura S, Ikegami A, Mizuno M, Yagi F, Nomura K. The expression profile of lectin differs from that of seed storage proteins in Castanea crenata trees., 2004, 68(8): 1698–1705.
[48] Ruiz KB, Biondi S, Martínez EA, Orsini F, Antognoni F, Jacobsen SE. Quinoa – a model crop for understanding salt-tolerance mechanisms in halophytes., 2016, 150(2): 357–371.
[49] Maughan PJ, Turner TB, Coleman CE, Elzinga DB, Jellen EN, Morales JA, Udall JA, Fairbanks DJ, Bonifacio A. Characterization of salt overly sensitive 1 () gene homoeologs in quinoa (Willd.)., 2009, 52(7): 647–657.
[50] Schmöckel SM, Lightfoot DJ, Razali R, Tester M, Jarvis DE. Identification of putative transmembrane proteins involved in salinity tolerance inby integrating physiological data, RNAseq, and SNP analyses., 2017, 8: 1023.
[51] Böhm J, Messerer M, Müller HM, Scholz-Starke J, Gradogna A, Scherzer S, Maierhofer T, Bazihizina N, Zhang H, Stigloher C, Ache P, Al-Rasheid KAS, Mayer KFX, Shabala S, Carpaneto A, Haberer G, Zhu JK, Hedrich R. Understanding the molecular basis of salt sequestration in epidermal bladder cells of., 2018, 28(19): 3075–3085.e7.
[52] Imamura T, Yasui Y, Koga H, Takagi H, Abe A, Nishizawa K, Mizuno N, Ohki S, Mizukoshi H, Mori M. A novel WD40-repeat protein involved in formation of epidermal bladder cells in the halophyte quinoa., 2020, 3(1): 513.
[53] Liu JX, Wang RM, Liu WY, Zhang HL, Guo YD, Wen RY. Genome-wide characterization of heat-shock protein 70s fromand expression analyses ofin response to drought stress., 2018, 9(2): 35.
[54] Golldack D, Li C, Mohan H, Probst N. Tolerance to drought and salt stress in plants: unraveling the signaling networks., 2014, 5: 151.
[55] Liu HC, Liao HT, Charng YY. The role of class A1 heat shock factors () in response to heat and other stresses in., 2011, 34(5): 738–751.
[56] Hinojosa L, Sanad MNME, Jarvis DE, Steel P, Murphy K, Smertenko A. Impact of heat and drought stress on peroxisome proliferation in quinoa., 2019, 99(6): 1144–1158.
[57] Jacobsen SE, Christiansen JL, Rasmussen J. Weed harrowing and inter-row hoeing in organic grown quinoa (Willd.)., 2010, 39(3): 223–227.
[58] Mestanza U, Riegel R, Silva H, Vásquez SC. Characterization of the acetohydroxyacid synthase multigene family in the tetraploide plant., 2015, 18(6): 393–398.
[59] Ceccato DV, Bertero HD, Batlla D. Environmental control of dormancy in quinoa () seeds: two potential genetic resources for pre-harvest sprouting tolerance., 2011, 21(2): 133–141.
[60] Xi WY, Liu C, Hou XL, Yu H. Mother of FT andregulates seed germination through a negative feedback loop modulating ABA signaling in., 2010, 22(6): 1733–1748.
[61] López-Marqués RL, Nørrevang AF, Ache P, Moog M, Visintainer D, Wendt T, Østerberg JT, Dockter C, Jørgensen ME, Salvador AT, Hedrich R, Gao CX, Jacobsen SE, Shabala S, Palmgren M. Prospects for the accelerated improvement of the resilient crop quinoa., 2020, 71(18): 5333–5347.
[62] Nakamura S, Pourkheirandish M, Morishige H, Kubo Y, Nakamura M, Ichimura K, Seo S, Kanamori H, Wu JZ, Ando T, Hensel G, Sameri M, Stein N, Sato K, Matsumoto T, Yano M, Komatsuda T. Mitogen-activated protein kinase kinase 3 regulates seed dormancy in barley., 2016, 26(6): 775–781.
[63] Danielsen S, Bonifacio A, Ames T. Diseases of quinoa ()., 2006, 19(1-2): 43–59.
[64] Khalifa W, Thabet M. Variation in downy mildew () resistance of some quinoa (Willd.) cultivars under Egyptian conditions., 2018, 7(2): 671–682.
[65] Mhada M, Ezzahiri B, Benlhabib O. Assessment of downy mildew resistance () in a quinoa (Willd.) germplasm., 2014, 8(3): 277–280.
[66] Ochoa J, Frinking HD, Jacobs T. Postulation of virulence groups and resistance factors in the quinoa/downy mildew pathosystem using material from Ecuador., 1999, 48(3): 425–430.
[67] Rasmussen C, Lagnaoui A, Esbjerg P. Advances in the knowledge of quinoa pests., 2003, 19(1-2): 61–75.
[68] Pereira E, Encina-Zelada C, Barros L, Gonzales-Barron U, Cadavez V, Ferreira ICFR. Chemical and nutritional characterization of quinoa (Willd.) grains: A good alternative to nutritious food., 2019, 280: 110–114.
[69] Ruiz KB, Biondi S, Oses R, Acuña-Rodríguez IS, Antognoni F, Martinez-Mosqueira EA, Coulibaly A, Canahua-Murillo A, Pinto M, Zurita-Silva A, Bazile D, Jacobsen SE, Molina-Montenegro MA. Quinoa biodiversity and sustainability for food security under climate change. A review., 2014, 34(2): 349–359.
[70] Zurita-Silva A, Fuentes F, Zamora P, Jacobsen SE, Schwember AR. Breeding quinoa (Willd.): potential and perspectives., 2014, 34(1): 13–30.
[71] Lin C, Liu ZJ, Dong YM, Michel V, Mao ZC. Domesticated cultivation and genetic breeding of., 2019, 41(11): 1009–1022.
林春, 刘正杰, 董玉梅, Michel Vales, 毛自朝. 藜麦的驯化栽培与遗传育种. 遗传, 2019, 41(11): 1009–1022.
[72] Ma XL, Zhang QY, Zhu QL, Liu W, Chen Y, Qiu R, Wang B, Yang ZF, Li HY, Lin YR, Xie YY, Shen RX, Chen SF, Wang Z, Chen YL, Guo JX, Chen LT, Zhao XC, Dong ZC, Liu YG. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in Monocot and Dicot plants., 2015, 8(8): 1274–1284.
Progress on genomics and locus of important agronomic traits in
Linghong Li1, Tong Gou1, Aixia Ren1, Pengcheng Ding1, Wen Lin1, Xiangyun Wu2, Min Sun1, Zhiqiang Gao1
Quinoa (, Willd.) as a new health food in the 20th century, its comprehensive nutritional composition, stress resistance and other characteristics have been paid much of attention, and enjoys the reputation of “nutritional gold”, “vegetarian king” and “food in the future” in the world. In recent years, with the rapid development of genomics and high-throughput sequencing technology, the high-quality whole genome sequence of quinoa has been completed, and the omics analysis and functional research of a series of key genes have been gradually carried out. In this review, we summarize the research progress in quinoa genomics, gene family analysis of important transcription factors, genetic map construction, QTL mapping of important traits, and genes for important agronomic and yield traits. Moreover, according to the current status of quinoa breeding, this paper also put forward five key problems in quinoa breeding, and pointed out four important directions of genetic improvement and breeding of quinoa in the future, so as to provide reference for the realization of directional genetic improvement of quinoa in the future.
quinoa; genomics; genetic breeding; traits; genes
2022-09-02;
2022-10-10;
2022-10-27
山西省基础研究计划项目(编号:202103021223158),山西省优秀博士来晋奖励项目(编号:SXBYKY2022022)和山西农业大学博士科研启动项目(编号:2021BQ82)资助[Supported by the Fundamental Research Program of Shanxi Province (No.202103021223158), the Scientific Research Project of Shanxi Province Outstanding Doctoral Work Award Fund (No.SXBYKY2022022), and the Doctoral research Project of Shanxi Agricultural University (No. 2021BQ82)]
李玲红,博士,讲师,研究方向:小麦和藜麦遗传育种。E-mail: lilinghong00en@163.com
孙敏,博士,教授,研究方向:小麦和藜麦栽培。E-mail: sm_sunmin@126.com
10.16288/j.yczz.22-289
(责任编委: 宿振起)
