头颈部鳞状细胞癌焦亡相关lncRNA对预后风险模型的构建与评估
2022-11-12罗倩詹雪冰况云舒陶香香王俊梁箫王俊杰孙恩涛陈冰
罗倩,詹雪冰,况云舒,陶香香,王俊,梁箫,王俊杰,孙恩涛,陈冰
(1. 皖南医学院病理教研室,安徽 芜湖 241002;2. 皖南医学院检验学院,安徽 芜湖 241002)
头颈部鳞状细胞癌(head and neck squamous cell carcinoma,HNSCC)是全球第六大最常见的恶性肿瘤,年发病率超过60万[1]。它通常与人类乳头瘤病毒(HPV)感染、酗酒或接触烟草致癌物有关[2]。目前手术、放疗和同步全身治疗技术在不断进步,但大多数HNSCC患者的早期临床症状不明显,发现时已处于中晚期,预后较差,且HNSCC患者的5年总生存率为50%[3-4]。因此,寻找有效的生物标志物和建立可靠的新的预后模型是十分必要的。
细胞焦亡(pyroptosis)又称细胞炎性坏死,是一种程序性细胞死亡,表现为细胞不断胀大直至细胞膜破裂,导致细胞内容物释放进而激活强烈的炎症反应[5]。据报道[6],细胞焦亡可被部分非编码RNA及其他分子调控进而影响肿瘤的增殖、侵袭和迁移。长链非编码RNA(long noncodingRNAs,lncRNAs)是转录长度超过200个核苷酸的非编码RNA,没有或几乎没有蛋白质编码能力,且在正常的生物环境和包括肿瘤发生在内的病理过程中,lncRNAs是调控靶基因不可或缺的[7-8]。REN N S等[9]发现lncRNAADAMTS9-AS2通过激活NLRP3介导的细胞焦亡,与miR-223-3p结合,抑制胃癌细胞增殖,提高顺铂敏感性。CHEN Z H等[10]发现,在肝癌中,NLRP3依赖的细胞焦亡通过lncRNA SNHG7/miR-34a/SIRT1轴被抑制。以上例子说明lncRNAs可作为肿瘤发生、发展的关键分子,通过直接或间接作用介导细胞焦亡。
为了寻找HNSCC新的治疗靶标,并准确预测HNSCC患者的预后,制定精准的治疗方案,本研究利用癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库[11]的转录组测序数据进行分析,构建了一种新的预后风险模型。但转录组测序数据是采用高通量基因测序技术得到的,而这一技术得到的基因表达数据有一定的局限性,即基因表达检测的平台和时间不同,对检测到的基因表达水平存在批次效应,可能导致分析结果不准确[12]。所以本研究采用了一种新的方法,以克服不同平台的批量效应。该方法是根据基因表达水平的相对排序对表达矩阵进行归一化和缩放[13],即将每个样本中的焦亡相关lncRNAs(prlncRNA)的基因表达水平进行两两比较并构建焦亡相关lncRNA对(PRLPs)。在某样本中,如果某PRLPs的第一个lncRNA的表达值大于第二个lncRNA,则该PRLPs在该样本中的得分为1;否则为0。计算所有样本中每个PRLPs的得分,剔除低变异性的PRLPs,即在任何数据集中,某一PRLPs的得分为1或0的样本低于20%,被剔除。最后,鉴定出具有较高变异性的PRLPs,以供进一步分析。该方法构建的预后风险模型具有临床实用性,能够区分临床病例的高低风险,且已被证明是可行的[14-15]。基于此,本研究从TCGA数据库获取HNSCC的转录组测序数据及临床资料,筛选出prlncRNA并以成对的形式显示,构建HNSCC预后PRLPs预后风险模型并进行评估,有望为HNSCC治疗策略的制定提供帮助。
1 材料和方法
1.1数据来源 HNSCC的转录组测序数据及临床资料于2021年5月18日从TCGA数据库(https://portal.gdc.cancer.gov/)下载。转录组数据共546例,包含44例正常头颈部组织和502例头颈部鳞状细胞癌组织。临床资料包括年龄、性别、生存时间、生存状态、肿瘤病理分期、肿瘤分级、TNM分期,剔除生存时间未知或小于30 d的患者肿瘤样本,纳入491例HNSCC样本,并以1∶1的比例随机分为训练集(246例)和验证集(245例)。
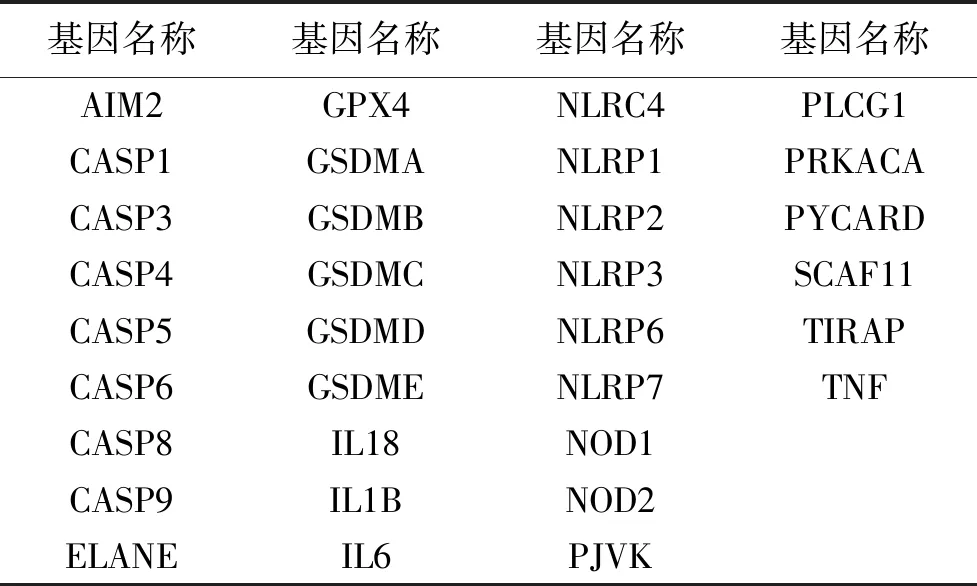
1.2数据预处理 使用perl语言脚本提取HNSCC的基因表达矩阵。从Ensembl (http://asia.ensembl.org)下载基因注释文件,将HNSCC基因表达矩阵中的ID转换为基因名称,并区分lncRNA和mRNA,获取HNSCC lncRNA表达矩阵。引用YE Y等[16]总结的33个焦亡相关基因,并采用与前述相同方法得到33个焦亡相关基因的HNSCC基因表达矩阵。使用perl语言脚本提取HNSCC的临床信息,用于后续研究分析。
1.3PRLPs的获取 利用R语言“limma”包,对HNSCC lncRNA表达矩阵与33个焦亡相关基因的HNSCC基因表达矩阵进行共表达分析,筛选出prlncRNA(相关系数Cor>0.4和P<0.001),并利用R包“igraph”可视化共表达关系,得到共表达网络。同时以FDR<0.05和logFC>2作为临界值对prlncRNA进行差异分析,得到差异表达的prlncRNA。为了解决数据批次校正问题,将所有差异prlncRNA以两两组合的形式进行分析,即lncRNA A | lncRNA B,称之为PRLPs。当lncRNA A的表达高于lncRNA B时,将PRLPs记为1;否则,记为0。得分为0或1的总数占所有样品的20%~80%,是有效组合。
1.4PRLPs预后风险模型的构建及评估 对PRLPs进行单因素Cox回归分析,得到与预后相关的PRLPs。再进行10次交叉验证的Lasso回归分析,在此之前需要进行Lasso回归过滤,使相对不显著的变量系数为0的PRLPs排除在建模之外,得到最小的误差值去构建Lasso回归预测模型,并采用分步法生成Cox比例风险模型。计算每个HNSCC样本的风险评分,并根据风险评分的中位数将HNSCC患者分为高、低风险组。风险评分大于中位数为高风险组;反之,则为低风险组。风险评分=coef1*Exp1+coef2*Exp2...+coefN*ExpN(Exp:某PRLPs比较之后的值;ef:回归系数)。计算所有样本的风险得分,并以散点图的形式显示出来。Kaplan-Meier分析显示高风险组与低风险组的预后情况。绘制ROC曲线评估PRLPs模型的灵敏度和特异度,其通过ROC曲线下方面积大小展示,即AUC值。进行单因素和多因素独立预后分析,并绘制多指标ROC曲线。对不同临床病理特征进行相关性分析,并以箱线图和热图的形式展现。以上使用了R语言“limma”、“survival”、“timeROC”、“caret”、“survminer”、“glmnet”和“survivalROC”包。
1.5风险评估模型与免疫检查点相关生物标志物之间的分析 免疫检查点相关的生物标志物有CTLA4、PDCD1、LAG3、HAVCR2和TIGIT。采用相关检验方法和差异分析方法对免疫检查点相关生物标志物进行相关性分析。分析免疫治疗与肿瘤微环境和干细胞指数的关系。采用免疫亚型分析来评估免疫治疗的疗效。上述分析用到了UCSC Xena数据库(https://xena.ucsc.edu/)的Subtype和StemnessScores数据。
2 结果
2.1差异prlncRNA的鉴定及PRLPs的建立 本研究的流程图,见图1。TCGA中HNSCC的详细临床特征,见表1。本研究用到的33个焦亡基因见表2。首先对HNSCC样本中的lncRNA和33个焦亡基因进行Person相关性分析(相关系数Cor>0.4和P<0.001),共鉴定出187个prlncRNA,结果见共表达网络图(见图2A)。随后以FDR<0.05和logFC>2作为临界值进行差异分析,得到53个差异prlncRNA(见图2B),其中4个下调,49个上调(见图2C)。最后对差异prlncRNA进行循环,以0.2 图1 研究流程图 2.2风险评估模型的建立 将HNSCC患者生存时间未知或小于30 d的样本删除,随后以1∶1的比例随机分为训练集(246例)和验证集(245例)。然后对上述筛选出的877对PRLPs进行单因素Cox回归分析,得到124个预后相关的PRLPs,其中60个PRLPs是高风险(HR>1,P<0.05),64个是低风险(HR<1,P<0.05)。再对预后相关的PRLPs进行Lasso回归分析得到最小误差值18,并计算相应回归系数(见图3A、图3B)。最后,利用上述得到的18个PRLPs构建Lasso回归预测模型,并采用分步法生成Cox比例风险模型(见图3C)。 表1 491例HNSCC患者临床病理特征 表2 33个焦亡基因 2.3风险评估模型的验证 在训练集(246例)和验证集(245例)中,根据训练集的风险评分的中位数将HNSCC患者分为高风险组和低风险组(见图4A、图4B)。结果发现低风险组患者预后更好(见图4C、图4D)。说明低风险组的临床治疗效果更好。随后,进一步通过1年、3年和5年ROC曲线评估PRLPs模型的灵敏度和特异度,在训练集中1年、3年和5年总生存AUC值分别为0.746、0.761、0.762(见图4E), 注:33个焦亡相关基因和lncRNA的共表达网络图(A);筛选出53个差异prlncRNAs的热图(B)和火山图(C)。 在验证集中1年、3年和5年总生存AUC值分别为0.715、0.660、0.660(见图4F)。表明PRLPs预后风险模型能较准确地预测HNSCC患者的生存预后。 2.4HNSCC患者的独立预后因素 为验证PRLPs预后风险模型是否可以独立于其他的临床性状作为独立的预后因子,进行单因素和多因素独立预后分析。图5A和图5B分别是训练集和验证集单因素和多因素独立预后分析的结果,结果表明模型风险评分与患者总生存时间相关(P<0.05)。多指标联合分析的ROC曲线结果表明训练集(见图5C)和验证集(见图5D)各自的1年总生存AUC值分别为0.746和0.715,均高于其他临床病理参数。以上结果表明,PRLPs预后风险模型可作为预测HNSCC患者预后的独立预后因素。 2.5风险评估模型与临床病理特征的关系 根据临床病理资料对HNSCC患者进行了分层分析。在年龄≤65岁、年龄>65岁、男性、G1-2、G3-4、T1-2、T3-4、M0、N0、N1-3和Ⅲ~Ⅳ期中, HNSCC高风险组患者的总生存时间较低风险组患者的总生存时间短(P<0.001),见图6。而女性、M1和Ⅰ~Ⅱ期这3个分层中,输出结果无统计学意义,说明所构建的风险模型只 A:Lasso回归系数分布的剖面图;B:10倍交叉验证选择最优λ值;C:分步法生成的Cox比例风险模型。 A、B为头颈部鳞状细胞癌中位风险评分的分布情况及风险评分与生存时间的关系,黑色虚线是把患者分为高风险组和低风险组的最佳分界线;C、D为头颈部鳞状细胞癌的生存曲线,红色代表高风险组,蓝色代表低风险组;E、F为预后模型的ROC曲线。 A、B为单因素和多因素Cox回归分析;C、D为多指标ROC曲线。 图6 头颈部鳞状细胞癌患者不同病理特征分层的高低风险生存分析 能在某些人群中适用,存在局限性。以上结果表明,PRLPs预后风险模型在不同分层中具有较好的预测能力。根据Wilcoxon符号秩检验计算不同临床病理特征之间的风险评分差异。结果显示,性别(见图7A)、分级(见图7B)、分期(见图7C)、T分期(见图7D)与风险评分存在显著相关性(P<0.05)。采用卡方检验来探讨HNSCC发病风险与临床病理特征之间的关系。结果如图7E所示。 2.6风险模型与免疫治疗的关系 免疫检查点相关生物标志物的相关性检验和差异分析结果分别如图8A和图8B所示,结果表明免疫检查点相关生物标志物的表达与风险评分存在显著相关性和差异性。图8C显示了HNSCC中免疫检查点相关生物标志物与DNA stemness score(DNAss)、RNA stemness score (RNAss)、StromalScore、ImmuneScore和ESTIMATEScore之间的相关性。免疫分型表明HNSCC中的CTLA4、LAG3、PDCD1、TIGIT和HAVCR2的表达在不同免疫亚型中存在明显差异,其中CTLA4、PDCD1、TIGIT和HAVCR2在C6中高表达,LAG3在C2中高表达(见图8D)。 A~D:Wilcoxon符号秩检验,性别(A)、分级(B)、分期(C)和T分期(S)与风险评分存在显著相关性(P<0.05);E:χ2检验,结果以热图展示(<0.01=**,<0.05=*)。 A:免疫检查点相关生物标志物的相关性检验;B:免疫检查点相关生物标志物的差异分析;C:免疫检查点相关生物标志物与DNAss、RNAss、Stromal Score、Immune Score和ESTIMATEScore之间的相关性;D:免疫检查点相关生物标志物的免疫分型。 随着高通量基因测序技术的发展和大规模基因表达数据集的建立,肿瘤研究人员能够准确识别与肿瘤预后相关的生物标志物[17]。但高通量基因测序技术得到的转录组测序数据有一定的局限性,即存在批次效应,可能会导致结果不准确[12]。因此,为了更精准地预测HNSCC患者的预后和疗效,本研究采用一种有效的新方法去构建预后风险模型。研究表明,在癌症预测模型的准确性方面,基因对组合形式优于单基因组合形式[12]。与传统的由单基因组成的预后模型不同,由基因对组成的预后模型不需要在不同患者基因表达矩阵测序平台上进行归一化处理,即:只需要在基因对的构建过程中考虑数据内基因的比较,而不需要对数据间的基因进行批量校正。因此本研究采用基因对去构建预后风险模型,以期更精准地预测肿瘤患者的预后和疗效。 细胞焦亡是一种程序性细胞死亡,由炎性小体诱导。研究表明,诱导肿瘤细胞焦亡去消灭肿瘤细胞在肿瘤治疗中尤为重要[18]。如在非小细胞肺癌中,转录因子p53通过促进肿瘤细胞焦亡抑制肿瘤生长[19];在肾细胞癌中,抑制BRD4可增强NLRP3炎症小体的转录活性,诱导细胞焦亡进而一直肿瘤细胞的增殖和上皮-间质转化[20]。目前研究主要从分子机制方面阐明细胞焦亡对肿瘤的影响,对肿瘤患者的预后研究仍较少,因此,利用细胞焦亡构建预后模型对于肿瘤患者的个性化治疗和预后预测具有重要意义。 LncRNAs是转录长度超过200个核苷酸的非编码RNA,没有或几乎没有蛋白质编码能力[7]。WANG B L等[21]研究表明在多种癌症类型中lncRNA-ATB的高表达是不良预后的标志,且标志着肿瘤患者淋巴结转移和神经浸润的高风险。ZHANG Y等[22]研究表明lncRNADSCAM-AS1可能作为一种新的预后标志物和潜在的治疗靶点,lncRNADSCAM-AS1主要是通过与YBX1相互作用,调节FOXA1和ERα的表达,促进肿瘤进展。以上研究表明,lncRNAs在肿瘤的发生、发展和预后中具有重要作用。 鉴于lncRNA和细胞焦亡在肿瘤的发生发展中都起着重要作用。因此,本研究利用生物信息学分析prlncRNA在HNSCC进展预后中的预测效能。首先对TCGA数据库中HNSCC lncRNA表达矩阵与33个焦亡基因的表达矩阵进行共表达分析,筛选出prlncRNA,进行差异表达分析,并以lncRNA对的形式进行展示,其中具有较高变异性的lncRNA对用于后续分析。然后,将HNSCC样本分为训练集和验证集,进行单因素Cox回归分析和Lasso回归分析,得到18个预后相关的PRLPs并构建预后风险模型。随后用中位风险评分区分高风险组和低风险组并进行生存分析,结果表明低风险组患者的总生存时间更长,表明其临床治疗效果更好,且训练集和验证集的AUC值均>0.7。说明所构建的PRLPs模型对于HNSCC患者的预后具有较好的预测效能。此外,采用单因素和多因素Cox回归分析及多指标联合分析的ROC曲线,研究发现PRLPs预后风险模型可以独立于其他的临床性状作为独立的预后因子。最后,据临床病理资料对HNSCC患者进行分层分析,发现PRLPs预后风险模型在不同分层中具有较好的预测能力。 此外,本研究还对风险评估模型和免疫检查点相关生物标志物之间的关系进行了联合分析。风险评估模型和免疫检查点相关生物标志物结果显示,高风险组中LAG3、PDCD1、TIGIT、CTLA4表达上调。在RNA或DNA水平上,免疫检查点相关的生物标志物也与干细胞指数评分呈负相关,与StromalScore、ImmuneScore和ESTIMATEScore呈正相关。此外,本研究还分析了免疫检查点相关生物标志物的免疫亚型:C1(伤口愈合型)、C2 (IFN-γ主控型)、C3(炎症型)、C4(淋巴细胞消减型)、C5(免疫静默型)和C6 (TGF-β主导型)[23]。C1、C2和C3型对免疫治疗敏感,若术前确定患者病理标本属于这3种亚型,可采用新辅助免疫治疗;若为C4、C5和C6型,则不适合新辅助免疫治疗术[23-25]。但是,免疫亚型仅仅适用于手术前,而手术后还需要ctDNA监测。ctDNA不仅是早期癌症的重要筛查指标,还可用于预测肿瘤患者术后病情的变化[26]。由于ctDNA是动态监测,因此收集HNSCC患者血液存在一定困难。 综上所述,本研究采用一种新颖的、可靠的方法构建了一个PRLPs预后风险模型,该模型可能为HNSCC患者早期诊断和预后提供新思路,并可能为HNSCC的预后诊疗提供新的方法和新的理论依据。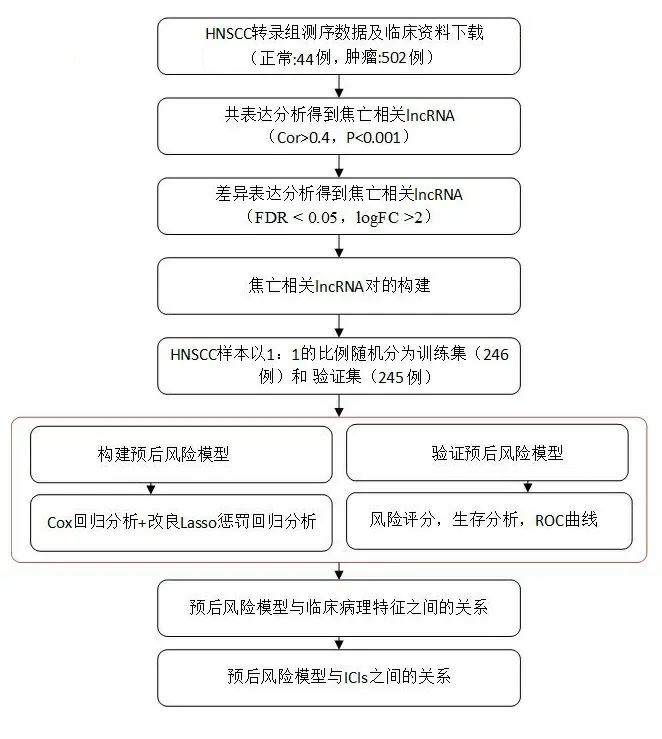












3 讨论
