煤直接液化催化剂预硫化过程中氢的作用研究
2022-11-07代浩杉熊严坤冯富祥
代浩杉,田 磊,熊严坤,冯富祥,杨 勇,3,马 智,郭 强,3,*,刘 源,*
(1.天津大学 化工学院,天津 300350;2.中科合成油技术有限公司 国家能源煤基液体燃料研发中心,北京 101407;3.中科合成油内蒙古有限公司,内蒙古 鄂尔多斯 010300)
中国富煤少油的能源结构决定了发展煤直接液化的必要性,煤直接液化过程通常都需要在较高的温度(约450 ℃)和压力(>10 MPa)下进行[1,2],主要包含较高温度下的热裂解和自由基加氢两个过程[3,4],热解产生的煤自由基需要被氢源(供氢溶剂和氢气)猝灭;若自由基不能及时被猝灭,自由基之间将会发生缩合反应生成大分子焦[5]。当氢气作为氢源时,氢气可以在催化剂上活化实现对自由基有效猝灭;当供氢溶剂作为氢源时,供氢溶剂会发生脱氢反应失去供氢能力,需要对脱氢的供氢溶剂进行催化加氢。基于以上两点,催化剂的加氢能力在煤液化过程中尤为重要[6]。
煤直接液化的催化剂可以分为三类[7],分别是钴钼镍系催化剂、酸性催化剂和铁基催化剂。钴钼镍系催化剂加氢能力强[8],但存在回收困难和环境不友好等问题;而酸性催化剂(例如ZnCl2)可以有效降低反应温度[9],但氯化物对设备的腐蚀限制其工业化应用;铁基催化剂由于其价格低廉、环境友好、加氢活性较高被广泛的研究,磁黄铁矿(Fe1-xS)是煤直接液化过程的活性相[7,10-12]。改善催化剂和煤的有效接触也可以有效的提高催化剂效率,Hu等[13]和Liu等[14]通过在煤上原位负载Fe2S3,发现煤转化率和油收率均有不同程度的升高。
从化学工艺角度来讲,铁基催化剂通常在煤液化过程的升温升压阶段在位硫化,实现催化剂的起活[15]。然而,在位硫化可能并非是最佳的催化剂活化条件;因此,有必要对催化剂前驱体进行适当的预硫化处理[16-19],从而实现催化剂活性的释放。催化剂前驱体的硫化预硫化可以分为干法硫化和湿法硫化[20],干法硫化更适用于温度敏感的催化剂。
煤是以稠环芳烃为基本结构单元,通过桥键相互连接的高分子有机化合物[21]。由于煤结构复杂含有较多杂原子,模型反应是研究煤直接液化反应的有力手段[22,23]。四氢萘作为最常见的模型供氢溶剂被广泛研究,可以通过萘加氢至四氢萘作为评价催化剂的活性的标准。
作者之前的工作[24]在5% H2S-N2的硫化气氛中研究了预硫化过程中硫的传递过程以及反应升温过程临氢催化剂的变化,实验证明,不同条件预硫化得到的催化剂在氢气的作用下均会发生转变。不难推测,催化剂前驱体一旦和硫化氢反应生成一部分硫化物后,预硫化过程中氢气会起到某些作用。但遗憾的是,之前的工作没有考虑氢气直接在预硫化过程中的影响。本研究在之前实验的基础上,在预硫化过程中引入氢气(5% H2SH2-N2),以探究氢气在典型的预硫化温度下的作用。
1 实验部分
1.1 试剂及催化剂前驱体制备
催化剂前驱体Fe2O3由共沉淀法制备,具体操作为将硝酸铁(Fe(NO3)3,99.9%,天津化工有限公司)、氨水和溶胶硅(SiO2)按照一定物质的量比加入三口烧瓶中,通过加入氨水调节pH值,在pH =9.0静置12 h进行共沉淀。沉淀完成后过滤,并使用去离子水洗涤三次。将上述洗涤后的滤饼加入去离子水高速剪切打浆,并在250 ℃下干燥喷雾,最后在350 ℃下焙烧5 h后得到催化剂前驱体—合成铁氧化合物。
1.2 催化剂前驱体的预硫化程序
将上述合成的催化剂前驱体在烘箱中120 ℃下烘干12 h,除去催化剂中吸收的水分,并将烘干后的催化剂置于干燥皿中备用。催化剂前驱体的预硫化过程在管式炉中进行,预硫化程序如下所述:称量2.00 g催化剂前驱体均匀放置于石英舟内,并将石英舟缓慢推入管式炉中部测温点,确保系统气密性后通入氮气置换系统中空气;置换完毕后,在氮气气氛中以10 ℃/min的升温速率升至指定温度,到达预硫化温度后切换至预硫化气氛停留1 h,气体流量500 mL/min,预硫化过程体系处于常压状态;预硫化完毕后,迅速将气氛切换至氮气,自然冷却1.0-2.0 h至室温,所制得的催化剂隔绝空气保存。其中,预硫化气氛为5% H2S-x%H2-N2(x=0、5、20、45、90、95),不同温度下预硫化得到的系列催化剂记为Cat-t-p,t为预硫化温度(单位:℃),p为预硫化过程中氢气分压(单位:%)。
1.3 催化剂加氢活性评价
对设备进行了标定矫正后,使用萘加氢反应来评价催化剂的加氢活性。上述预硫化后得到的2.00 g催化剂经正构烷烃C14液封后,转移至100 mL浆态床反应釜内,加入25.00 g萘后摇匀后密封反应釜。确保系统不漏气后,使用1% H2SH2的反应气氛置换管线中空气后背压至5 MPa,反应气体流量为200 mL/min;升温程序为从室温快速升温至200 ℃(此时系统压力已经达到5 MPa),200 ℃以2.25 ℃/min升温至300 ℃,300 ℃以1 ℃/min升温至360 ℃,在360 ℃停留1 h;使用磁力搅拌,反应过程控制转速为1200 r/min,反应装置如图1所示。
反应完毕,迅速摇下加热套并使用电风扇对反应釜降温,待降至室温后,停止进气并卸压。将冷阱、热阱以及反应釜中的产物收集摇匀,加入一定量的1,2,3,4-四氢喹啉作为内标物,通过内标法确定反应后萘的质量,并通过下式计算萘的转化率:
式中,α为萘转化率(单位:%),C0为反应前加入萘的质量(单位:g),Ci为反应后通过色谱内标计算萘的质量(单位:g),以萘转化率的高低反映催化剂加氢活性的强弱。
1.4 催化剂的表征
程序升温硫化和程序升温还原均采用实验室自搭管式炉,程序升温硫化采用安捷伦7890A气相色谱仪离线分析,硫化气氛为5% H2S-N2,升温速率2 ℃/min;程序升温还原使用质谱分析尾气,还原气氛为10% H2-Ar,升温速率5 ℃/min。
使用德国Bruker公司D8型X射线衍射仪对催化剂进行研究,工作电压为40 kV,工作电流为35 mA,扫描角度为10°-90°,步长0.025°,停留时间0.03 s。
Mössbauer谱图(MES)采用美国Austin公司S 2600 Mössbauer谱仪进行测试,检测过程采用等加速方式低温下测样,放射源为 25 m Ci 的 Co(Pd),测样采用 α-Fe 标定。基于早期课题组[24]的文献调研,将这些物种在 Mössbauer谱中的参数列于表1中,使用 Mösswinn 4.0 软件进行解谱分析,将参数输入软件后,即可拟合并得到拟合谱图与相应物质位点参数及其面积占比(计算所得物质含量存在 ± 5% 的误差),计算拟合结果可以反映各物种的相对含量。

表1 Mössbauer谱的拟合参数Table 1 Parameters of Mössbauer spectrum
硫铁比使用安东帕的Multiwave PRO和美国的PE 2100 ICP-OES测量所得,具体操作为准确称量适量催化剂,按照3:1的比例加入浓盐酸和浓硝酸,在微波消解仪上消解;消解过程在密闭体系中进行,以保证硫的测量的准确性;消解完成冷却至室温,转移至100 mL容量瓶中定容,由等离子发射光谱仪测定。
GC-MS采用安捷伦5973N测试,色谱柱为HP-5,升温程序从50 ℃以3 ℃/min升温至140 ℃,温度升至140 ℃后以5 ℃/min升温至300 ℃。
XPS 分析采用中科合成油技术有限公司分析测试平台的美国FEI 公司ESCALAB 250xi 型 X 射线光电子能谱仪测量Fe 2p3/2和S 2p轨道电子结合能。拟合参数为:Fe(0)的2p3/2结合能为706.3 eV,Fe(Ⅱ)-S的2p3/2结合能为707.6 eV,Fe(Ⅲ)-S的2p3/2结合能为708.6 eV,Fe(Ⅱ)-O的2p3/2结合能为710.0 eV,Fe(Ⅲ)-O的2p3/2结合能为714.0 eV;S 2p1/2= S 2p3/2+1eV,S2-的2p3/2结合能为161.3 eV,的2p3/2结合能为162.5 eV,S/S8的2p3/2结合能为163.4 eV。
SEM分析采用美国 FEI 公司 Quanta 400F型扫描电子显微镜。
2 结果与讨论
2.1 前驱体硫化温度的选取和催化加氢活性
2.1.1 催化剂前驱体的 TPS
对催化剂前驱体进行了程序升温硫化(TPS)测试,硫化气氛为5% H2S-N2,使用TCD检测器检测尾气中H2和H2S含量,其结果如图2所示。在35 ℃下,前驱体接触硫化氢130 min内尾气没有H2S信号,表明室温硫化过程足以发生,室温对于硫化过程并非是“低温”,硫化过程在室温下就相当剧烈。随后在此气氛下进行程序升温,升温阶段速率为2 ℃/min,发现催化剂前驱体在150 ℃后有H2S的二次消耗峰,前驱体与H2S二次反应的温度为150-500 ℃,并且在160-500 ℃出现H2的生成峰,表明程序升温至150 ℃后,不含氢硫化可以产生H2。不难推测,H2S可以在催化剂的表面上催化分解,释放H2。当温度升至640 ℃后,尾气中H2S信号急剧减弱,H2信号急剧上升,这可以归因于H2S的热分解。
基于以上对催化剂前驱体的认识,分别选取50、150和300 ℃作为典型的预硫化温度,探究不同温度下氢气在预硫化过程中的影响。
2.1.2 催化剂前驱体的 H2-TPR
催化剂前驱体的H2-TPR结果如图3所示,还原气氛为10%H2-Ar混合气,升温速率为5 ℃/min,使用质谱检测器检测尾气中H2的消耗。不难发现,催化剂前驱体的初始还原温度为160 ℃,表明氢气在160 ℃以下单独作用前驱体是惰性的;催化剂前驱体具有两个主要的氢消耗峰,分别可以对应于Fe2O3还原至Fe3O4(> 160 ℃)和Fe3O4还原至FeO(> 350 ℃)的过程。
2.1.3 不同温度下氢气分压对催化剂加氢活性的影响
氧化物前驱体、50、150和300 ℃不同氢气分压下预硫化1 h的催化剂的萘加氢转化率如图4所示。可以观察到,氧化物前驱体单独使用的活性很低,转化率不足4%;经过不同条件预硫化后,转化率明显提高,表明预硫化处理可以使催化剂有效活化。从图4中可以发现,50 ℃预硫化时,随着气氛中氢气分压的增大,活性呈现上升的趋势,由47.3%增加至56.3%,均高于氮气预硫化。150 ℃预硫化时,加氢活性同样随着氢气分压的增加而增大,由60.7%增加至69.1%;且由于硫化速率的增加,整体活性高于50 ℃预硫化。预硫化温度为300 ℃时,氢气的引入使得催化活性有不同程度的下降,均低于氮气预硫化过程,表明在不同温度下氢气在预硫化的过程作用不尽相同。同时,300 ℃预硫化整体活性介于150 ℃和50 ℃之间,表明适当硫化速率的选取在氧化物前驱体干法预硫化是非常重要的。
2.2 不同硫化温度下氢气分压对催化剂物相的影响
2.2.1 50 ℃硫化时催化剂物相分析
2.2.1.1 XRD与Mössbauer分析
50 ℃下,不同氢气分压下预硫化催化剂的XRD如图5所示。Cat-50的X射线衍射结果普遍弥散,表明该温度处理所得催化剂的晶粒小。随着氢气分压增大,归属于Fe2O3的衍射信号(41.621°和74.616°,PDF#39-1346)逐渐减弱,Fe3S4的特征衍射信号(2θ=34.935°、42.463°和61.635°,PDF#16-0713)明显增强,表明50 ℃预硫化氢气的引入可以加速硫化,有助于Fe3S4的生成。
Cat-50-p系列催化剂的Mössbauer谱拟合结果如表2所示,随着预硫化过程氢气分压的增大,硫化过程明显加快,催化剂中氧化物前驱体含量减少,硫化物种和单质Fe含量显著上升。其中,Fe3S4的含量由3.7%升至16.3%,这与XRD结果所一致;Fe1-xS物种的含量也由15.1%升至31.7%,但在XRD中并未观察到,表明Fe1-xS处于高分散的状态;Fe3O4的含量随着氢气的含量略有下降且含量较低,表明Fe3O4可能是硫化过程的中间态;Cat-50-0催化剂中氧化物总含量可高达62.1%,单质Fe含量为8.7%;而在Cat-50-95催化剂中氧化物总含量降至15.3%,单质Fe含量升至24.9%,单质Fe也处于高分散状态。此外,在50 ℃不同氢分压预硫化后的催化剂中均观察到单质S的生成。

表2 Cat-50-p的Mössbauer谱解析Table 2 Mössbauer spectrum results of Cat-50-p
2.2.1.2 GC-MS和ICP分析
为进一步确定硫化过程生成的单质硫的质量,对Cat-50-p使用THF洗涤溶解,加入一定量C14作为内标物超声后离心,取得上清液。将上述上清液,进行GC-MS分析测试通过内标法计算单质硫的质量,其GC-MS结果见图6,计算结果见表3,S质量的单位为g/gcat。对Cat-50-p系列催化剂进行微波消解,测定催化剂中铁和硫含量,进一步确定催化剂的S/Fe原子比,其结果见表3所示。由表3可以看到,50 ℃处理所得的催化剂的硫铁比随着氢气分压的增大呈现上升趋势,由0.87升至1.16,同样表明预硫化过程氢气的引入可以加速硫化;而随着预硫化过程氢气分压的增大,每克催化剂中单质S的质量从0.0155 g降至0.0056 g。Cat-50-p催化剂随着p值的增大,硫铁比越高,整体的单质S的含量越低。

表3 Cat-50-p的(S/Fe)atom和单质硫质量Table 3 (S/Fe) atomand elemental sulfur quality results for Cat-50-p
2.2.1.3 XPS分析
为了了解催化剂表面性质,选取 Cat-50-0、Cat-50-20、Cat-50-45和Cat-50-95进行XPS测试,拟合结果见表4。由表4可知,以上催化剂表面均没有观察到单质Fe,表明单质Fe位于颗粒的内部。随着p值的增大,表面的Fe-O键减少,Fe-S键从3.2%增至55.3%,这和体相规律一致,表明预硫化过程中氢气的引入有助于硫化过程。其中,Fe(Ⅱ)-O的出现也表明Fe3O4可能是硫化过程的中间态。在Cat-50-p表面观察到S/S8和,分别对应着单质硫和FeS2。随着p值的增大,催化剂中S/S8的含量下降,这和上述GC-MS计算结果一致;而S2-表面含量下降,催化剂整体硫化程度升高,表明S2-向体相传递,预硫化过程氢气的引入有利于硫的传递。
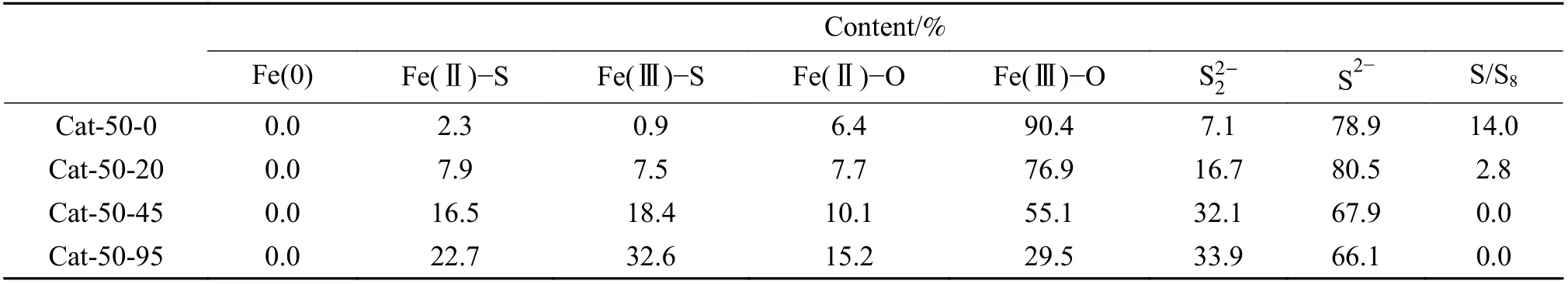
表4 Cat-50-p的XPS拟合Table 4 XPS fitting results of Cat-50-p
2.2.1.4 硫化结果讨论
结合上述表征结果,硫化预硫化过程是硫化氢在氧化铁颗粒表面进行化学吸附,反应生成某种硫化物(FexSy)。这些硫化物既可以解离吸附硫化氢,也可以解离吸附氢气,从而产生活性氢物种,预硫化过程氢气分压越大,表面产生的活性氢浓度越大。相比于硫,活性氢更易向催化剂颗粒内部传递,进入颗粒内部的活性氢与氧化物发生反应生成水留下空位,使表面的硫(、S2-或单质S)更易传递到颗粒内部,从而实现加速硫化。但由于50 ℃硫在催化剂内部传递较慢,该温度下存在大量的单质Fe,最终形成图7的结构。
2.2.2 150 ℃硫化时催化剂物相分析
2.2.2.1 XRD与Mössbauer分析
150 ℃不同氢气分压预硫化的XRD如图8所示。从图8中主要可以观察到三类衍射峰,分别归 属 于FeS2(38.612°、43.368°和55.660°,PDF#42-1340)、Fe3S4(34.935°和61.635°,PDF#16-0713)和Fe1-xS(34.838°、39.439°、51.178°和62.509°,PDF#29-0726),低氢气分压预硫化所得催化剂的衍射信号归属于FeS2,高氢气分压预硫化所得催化剂的衍射信号分别可以归属于FeS2、Fe3S4和Fe1-xS。随着预硫化气氛中氢气分压的增大,FeS2的衍射峰强度几乎不受影响,而Fe3S4和Fe1-xS的衍射峰强度逐渐增强,这表明预硫化过程氢气的引入加速硫化,有利于Fe3S4和Fe1-xS的生成。
Cat-150-p的Mössbauer谱拟合结果见表5所示。类似的,随着预硫化氢气分压的增大,催化剂中氧化物前驱体含量减少,硫化物种和单质Fe含量上升。其中,Fe1-xS的含量从23.9%升至43.8%,这和XRD结果所一致;在Cat-150-95催化剂中氧化物含量可降至7.1%,单质Fe含量升至10.4%,均低于Cat-50-95。此外,在150 ℃预硫化过程中均观察到FeS2存在,且随着氢气分压的增大其含量略有下降;150 ℃预硫化过程中没有观察到单质S的生成。

表5 Cat-150-p的Mössbauer谱解析Table 5 Mössbauer spectrum results of Cat-150-p
2.2.2.2 ICP分析
类似的,通过ICP分析进一步确定催化剂的S/Fe原子比,其结果见表6所示。可以看到150 ℃预硫化所得的催化剂的硫铁比随着氢气分压的增大呈现上升趋势,由0.92升至1.23,这和上述表征结果所一致。相比于50 ℃,150 ℃所得催化剂具有更高的S/Fe,且没有观察到单质S的生成,表明温度升高有利于硫的传递过程。

表6 Cat-150-p的(S/Fe)atomTable 6 (S/Fe) atomresults for Cat-150-p
2.2.2.3 XPS分析
选取 Cat-150-0、Cat-150-20、Cat-150-45和Cat-150-95进行XPS测试,拟合结果见表7。随着分压值的增大,表面的Fe-O键减少,Fe-S键从30.5%增至52.0%,与体相结果所一致,预硫化过程中氢气的引入可以加速硫化。但150 ℃低氢分压预硫化表面的Fe-O键含量明显低于50 ℃,这表明温度升高有利于硫的传递。

表7 Cat-150-p的XPS拟合结果Table 7 XPS fitting results of Cat-150-p
2.2.3 300 ℃硫化时催化剂物相分析
2.2.3.1 XRD与Mössbauer分析
Cat-300-p催化剂的XRD如图9所示,从图9中观察到三类衍射峰,分别归属于FeS2(38.612°、43.368°和55.660°,PDF#42-1340)、Fe1-xS(34.838°、39.439°、51.178°和62.509°,PDF#29-0726)和Fe3O4(2θ=41.375°和67.227°,PDF#19-0629)。在Cat-300-0催化剂主要观察到FeS2的衍射信号,伴随着部分Fe1-xS的衍射信号;在p≠0时,催化剂中主要观察到Fe1-xS的衍射信号,还观察到Fe3O4的衍射峰。
Cat-300-p的Mössbauer谱拟合结果见表8所示。由表8可知,Cat-300-0催化剂中主要得到FeS2和Fe1-xS物种,而p≠0时催化剂中FeS2含量明显下降,并且Fe1-xS系列物种含量显著上升,这和XRD结果所一致。随着预硫化过程氢气分压的增大,Cat-300系列催化剂中氧化物含量从41.1%降至22.5%,单质Fe的含量基本保持不变,最高可达3.9%,表明该温度预硫化氢气的引入有利于氧化物向硫化物(Fe1-xS)的转变。此外,在p≠0的Mössbauer谱拟合结果中观察到一定含量的Fe3O4。

表8 Cat-300-p的Mossbauer谱解析Table 8 Mössbauer spectrum results of Cat-300-p
2.2.3.2 ICP分析
通过ICP分析进一步确定Cat-300-p催化剂的S/Fe原子比,其结果见表9所示。可以看到300 ℃预硫化所得的催化剂的硫铁原子比,在p=0时硫铁原子为0.95,随着氢气分压的增大略有下降。

表9 Cat-300-p的(S/Fe)atom结果Table 9 (S/Fe) atomresults for Cat-300-p
300 ℃给出的硫铁原子比低于50和150 ℃,表明300 ℃预硫化存在硫化程度“不足”的现象。结合上述XRD、穆斯堡尔谱观察到Fe3O4的存在,以及前驱体H2-TPR的结果,推测300 ℃下p≠0预硫化产生的Fe3O4位于催化剂颗粒的内部,被外部的Fe1-xS包裹形成“核壳”结构,这种“核壳”结构阻碍硫的传递,导致催化剂颗粒硫化程度较低。
2.2.3.3 SEM-EDS分析
为了验证上述“核壳”结构的猜想,选取Cat-300-95催化剂为例。将Cat-300-95催化剂进行切割,并且在切面上选取不同的位置进行能谱测试(如图10所示),进而确定颗粒中O和S的含量(如表10所示)。从表10可知,催化剂颗粒中外层氧含量更低、硫含量更高;而靠近中心位置具有更高的O/S质量比,SEM-EDS结果证明了催化剂颗粒内部硫化深度更低,氧化物含量更高。值得注意的是,催化剂颗粒中O/S最高的位置并不一定处于颗粒的中心,不同颗粒中的O/S也不尽相同,这可能是由于催化剂颗粒的球心不在切面上,但总体而言催化剂颗粒外围的氧含量更低,内部氧含量更高,这样的结果证明300 ℃预硫化过程氢气的引入会形成硫化物包裹氧化物的“核壳”结构。

表10 Cat-300-95的SEM-EDS的(O/S)wtTable 10 (O/S) wtresults of SEM-EDS of Cat-300-95
2.2.3.4 XPS分析
300 ℃下分压为0、20、45和90时预硫化所得催化剂的XPS拟合结果见表11。类似的,在Cat-300-p系列催化剂中,随着预硫化气氛中氢气分压的增大,催化剂中Fe-S含量从36.8%增加至55.0%。值得注意的是,随着氢气分压的增大,Cat-300-p系列催化剂中S2-的含量明显增大。

表11 Cat-300-p的XPS拟合Table 11 XPS fitting results of Cat-300-p
2.3 不同温度典型气氛下的物相分析
选取不同温度(50、150和300 ℃)下p= 0和p= 95%预硫化所得催化剂,对其Mössbauer拟合结果进行比较,如表12所示。数据表明,50 ℃预硫化所得催化剂中没有观察到FeS2的生成,而在150和300 ℃预硫化中可以观察到,表明FeS2的生成对应着程序升温硫化(TPS)中的二次反应。氢气的引入均有利于氧化物向硫化物的转变,氧化物含量的下降程度为:150>300>50 ℃;预硫化过程氢气的引入,导致单质铁含量有不同程度的上升,单质铁含量上升程度为:50>150>300 ℃。

表12 不同温度下分压为0和95时的Mössbauer谱解析Table 12 Mössbauer spectrum results of 0 and 95 in different temperature
不同温度(50、150和300 ℃)下分压为0和95%时预硫化所得催化剂的硫铁比结果如表13所示。分压为0时,硫铁比随着温度升高而升高;而分压为95%时,Cat-150-95催化剂具有最高的硫铁比,300 ℃下氢气的引入使催化剂的硫铁比有一定程度的下降。值得注意的是,300 ℃预硫化氢气的引入使得加氢活性有一定程度的下降(见图4),表明300 ℃预硫化氢气引入的作用有所不同。

表13 不同温度下分压为0和95时的(S/Fe)atom结果Table 13 (S/Fe) atomresults for 0 and 95 in different temperature
不同温度(50、150和300 ℃)下分压为0和95%时预硫化所得催化剂的XPS拟合结果如表14所示。数据表明,不同条件下硫化所得催化剂表面均没有观察到单质Fe的存在。此外,Cat-50-0催化剂中能观察到大量的Fe-O和S/S8,随着温度的升高,Fe-O和S/S8的含量明显下降。分压为95%时,不同温度下Fe-S含量基本一致,但300 ℃预硫化所得S2-含量明显高于50和150 ℃,这就表明300 ℃预硫化时氢气可以将表面的硫还原至低价态,而S2-难以向内部传递。
综上所述,在50和150 ℃预硫化过程氢气的引入促进了氧化物前驱体向硫化物的转变。相比50 ℃,150 ℃硫化所得催化剂中观察到FeS2的生成,氧化物和单质Fe含量更低,Fe3S4和Fe1-xS的含量更高。在前驱体的TPS中,150 ℃有额外的H2S消耗峰,这种“额外”硫化过程可能对应150 ℃中FeS2的生成;Cat-150-p催化剂的硫铁原子比均高于Cat-50-p同气氛处理所得催化剂,在Cat-150-p的系列催化剂中没有检测到单质S的生成。预硫化过程氢气的引入,加速了硫的传递(如图7);升高温度硫传递更快,Cat-150-p中硫化物含量更多,氧化物和单质Fe含量更低,故整体活性高于Cat-50-p。

表14 不同温度下分压为0和95时的XPS拟合Table 14 XPS fitting results of 0 and 95 in different temperature
而Cat-300-p催化剂有较好的晶型。p=0时,有利于FeS2和Fe1-xS物种的生成;p≠0时,有利于Fe1-xS物种的生成。预硫化过程中氢气的引入有利于氧化物的硫化生成Fe1-xS,同时可以将催化剂颗粒内部的的Fe2O3还原至Fe3O4。过强的还原能力导致催化剂表面的硫还原至低价态,表面的S2-的含量较多,不利于实现向催化剂颗粒内部的硫传递过程,形成硫化物包裹氧化物的“核壳”结构,具有较低的硫铁原子比,进而随着氢气的引入催化活性有不同程度的下降。
3 结 论
氧化物前驱体的预硫化可以有效的活化催化剂,增强其萘加氢活性。
在不同温度下氢气的引入促进可以硫化,均有利于活性相Fe1-xS生成,但不同温度预硫化时,对催化剂物相结构与加氢活性的影响规律并不相同。50 ℃预硫化时,氢气的引入,在硫化物的作用下增强体系的还原能力,有利于硫的传递;随着氢气分压的增大,活性也随之提高,由47.3%提高至56.3%,但由于温度较低,硫传递过程较慢,整体活性较差。当升高温度至150 ℃,硫的传递加快,整体活性更高;随着氢气分压的增大,萘转化率由60.6%提高至69.1%。而300 ℃预硫化,氢气将表面硫化物还原至Fe1-xS不利于硫的传递,导致硫化深度不足,故随着氢气的引入活性有不同程度的降低。
预硫化过程氢气的作用机制是,氢气在硫化物表面活化,提高了体系中活性氢的浓度,这些活性氢可以向催化剂颗粒内部传递,将内部氧化物部分还原促进硫的传递,从而实现加速硫化;而温度过高还原能力增强,不利于硫的传递。
