基于RAD高通量测序的蓝花楹群体遗传分析
2022-10-17刘学锋李小梅张国武张沛健刘果黄敏陈银霞
刘学锋, 李小梅, 张国武*, 张沛健 , 刘果, 黄敏,陈银霞
基于RAD高通量测序的蓝花楹群体遗传分析
刘学锋1, 李小梅2, 张国武1*, 张沛健1, 刘果1, 黄敏1,陈银霞3
(1. 中国林业科学研究院速生树木研究所,广东 湛江 524022;2. 湛江科技学院,广东 湛江 524000;3. 江西省林业科技推广和宣传教育中心,南昌 330033)
为了解蓝花楹()种质资源的遗传多样性和群体遗传结构,对168份种质材料进行RAD-seq测序,构建了系统进化树并进行主成分、群体结构和遗传多样性分析。结果表明,比对参考基因组平均比对率为81.02%,平均测序深度23.18×,最终获得45 552个高质量的SNPs。群体遗传结构分析表明,供试蓝花楹可划分为2个大的类群,来自川、渝地区的种质材料基本归为一类;其余地区归为另一类。19个地区的蓝花楹在SNP水平上的遗传多样性较高,云南昆明(YNKM)居群的核苷酸多样性(π)和期望杂合度()最大,表现出最高的遗传多样性。因此,来自川、渝地区的蓝花楹具有相对较近的亲缘关系,推断来自同一祖先,而其余地区的种质可能是随机引种栽培。
蓝花楹;RAD-seq;遗传多样性;群体遗传
蓝花楹()是紫葳科(Bigno- niaceae)蓝花楹属植物,落叶乔木,原产南美洲,广泛分布于澳大利亚、南非、秘鲁、墨西哥、巴西、阿根廷等国,我国引种栽培于广东、广西、四川、福建和云南等地[1]。蓝花楹树形优美,蓝紫色花, 可作行道树,具有观花、观叶、观形等观赏价值,是一种值得推广应用的园林树种[2–3]。
对蓝花楹的研究主要集中在潜在栽培区预测、与木霉真菌的相互作用[4]、药用成分分析[5]和土壤重金属及化合物吸附[6–7]等方面。迄今为止,蓝花楹在分子水平上的研究仅限于染色体[8–9]、质体序列[10]、ISSR分子标记[11–12]等。因蓝花楹早期引种栽培的品种来源未知,种源关系混乱,严重影响了蓝花楹的种质资源保护、良种选育及创新利用,因此亟待新的方法来揭示国内蓝花楹的遗传多样性和群体遗传结构。近年来,随着基因组测序技术的飞速发展,为种质资源鉴定及遗传多样性研究提供了新的方法。限制性酶切位点关联DNA测序技术(restriction-site-associated DNA sequencing, RAD-seq)是在二代测序基础上发展起来的一种基于全基因组酶切位点的简化基因组测序技术。随着高通量测序技术和生物信息技术的发展,RAD-seq分析也日趋完善,并在很多生物上得以应用[13–15]。RAD-seq不仅可以得到可靠的高密度遗传标记,而且适用于没有参考基因组的物种研究[16]。Premarathne等[17]利用RAD-seq技术对来自不同地区的93份日本胡椒()种质资源进行了分析。Feng 等[18]利用RAD-seq技术对甘薯()的全基因组遗传多样性进行检测并分析群体结构,开发了相应的SSR标记。Fukuda等[19]利用RAD-seq 技术生成的SNP标记构建了枇杷()高密度遗传连锁图谱,可用于农艺性状的QTLs研究。黄承玲等[20]基于RAD-seq测序技术对贵州百里杜鹃保护区杜鹃花属()植物进行了分类,证实RAD-seq技术在复杂植物类群的物种分类方面比传统分子标记具有明显优势。
群体遗传学是研究植物群体遗传结构及其变化规律的遗传学重要学科。因此,本研究以在国内收集的168份蓝花楹种质为材料,利用RAD-seq技术进行蓝花楹的群体遗传分析,从基因组水平上揭示不同种质间的遗传分化关系,为蓝花楹种质资源的保存利用、育种策略的实施、分子标记辅助育种和关联图谱的构建提供理论依据和实践参考。
1 材料和方法
1.1 试验材料
分别从四川、重庆、福建、江西、云南、广东、广西7省(区、直辖市)的19个地区早期引种的蓝花楹()树上采集种子(表1), 每个自然居群选择9~16棵植株,相邻植株间隔约50 m,个体在10株以下的居群全部采样。福建莆田(1)和云南文山(1)居群为大树,福建莆田(2)和云南文山(2)居群为小苗,且相隔50 km以上。种子在位于广东湛江的南方国家级林木种苗示范基地分产地播种培育成1 a生小苗,于2018年6月从每个产地小苗中随机选取8株,剪取其新鲜健康的幼叶适量, 分别装入5 mL离心管中,再放入液氮速冻后带回, 编号并保存在–80 ℃冰箱备用。
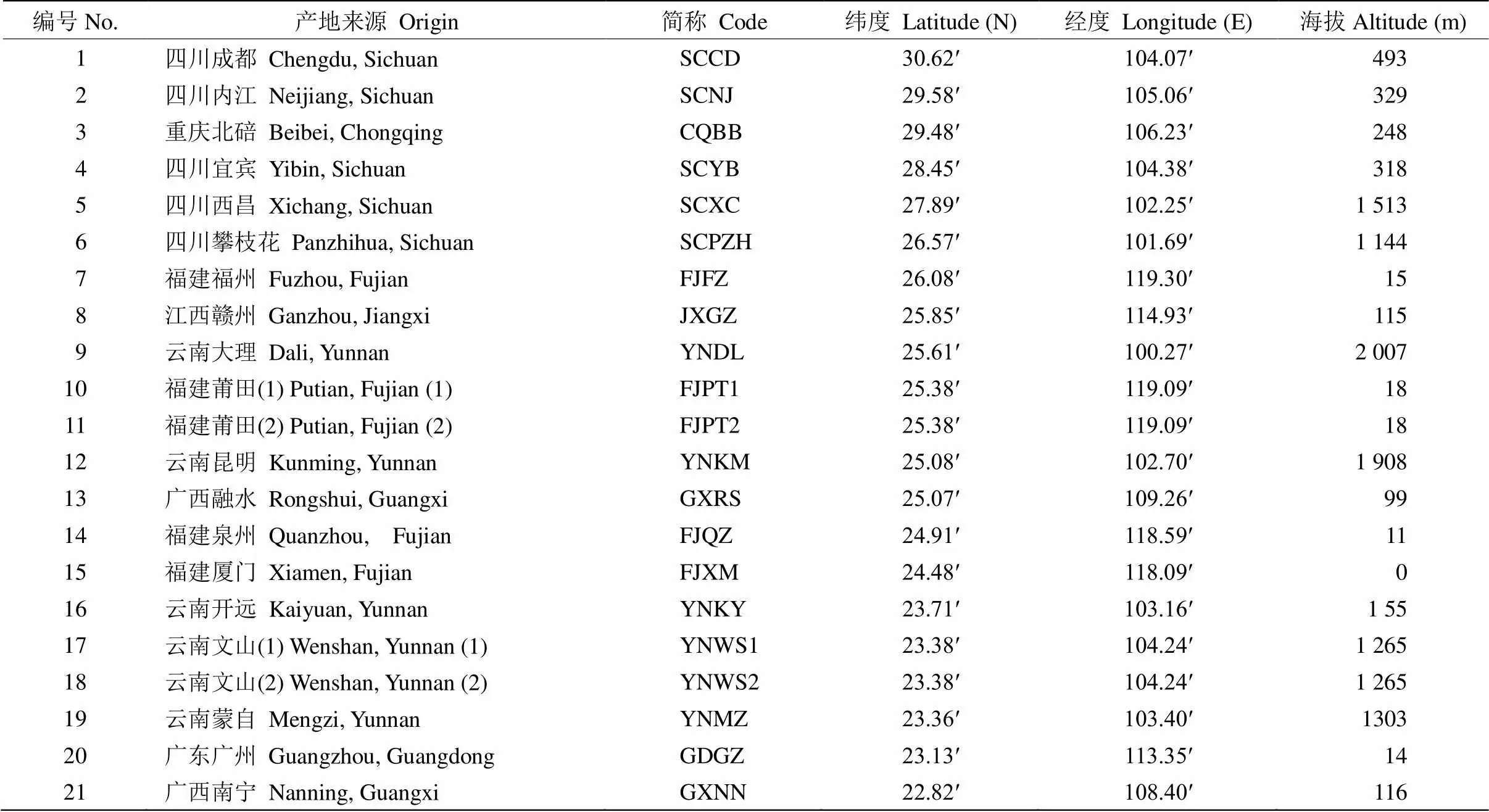
表1 蓝花楹试验材料及其来源
1.2 基因组DNA提取
采用CTAB法提取样本基因组DNA并进行质量检测,对质量合格的DNA进行下一步上机测序。
1.3 RAD文库构建
首先应用限制性内切酶R I对基因组进行酶切,然后每个样本分别进行物理打碎,选取200~ 400 bp插入片段文库。利用Illumina HiSeq2000测序平台进行双末端(Paired End-150)测序,所有的文库构建及上机测序都由北京诺禾致源科技股份有限公司完成。
1.4 测序reads过滤
对原始数据进行过滤,首先去除带接头(adapter)的reads pair;当单端测序read中含有的N含量超过该条read长度的10%时,需要过滤paired reads;当单端测序read中含有的低质量(Q≤5)碱基数超过该条read长度的50%时,去除此对paired reads[21]。
1.5 比对和变异检测
将组装样本带有酶切识别序列的reads进行聚类,并按照深度进行降序排序。将深度高的reads作为种子进行聚类。根据深度信息对聚类后的reads进行纠错、过滤重复区域等。根据聚类的结果,将一端的reads进行contig拼接,结合insert的大小和overlap关系,将拼接后的contig与聚集在另一端的reads连接起来,组装成最终的contig序列。使用VelvetOpti- miser软件对个体RAD局部组装,得到最后的组装序列,并对100 bp以下的contig进行过滤得到基因组[22]。基因组大小为82 851 747 bp,群体样本比对率为78.21%~83.46%,基因组平均测序深度为23.18×, 平均覆盖度为92.64%。有效的高质量测序数据通过BWA软件[23](参数:mem-t4-k32-M)比对到蓝花楹组装后的参考基因组,并过滤掉无法匹配和匹配到多处位点的序列;比对结果经SAM-TOOLS[24]去除重复(参数:rmdup)并归类[25]用于变异检测。经HWB (Hardy-Weinberg equilibrium)过滤后进行HWB检验,<0.05为遗传不平衡,需要过滤掉,软件:无,脚本执行。再进行LD (linkage disequilirium)过滤,连锁强度2>0.8, 软件:plink1.9–allow-extra-chr–indep- pairwise 50 5 0.8。
1.6 数据的统计分析
群体遗传结构分析 经检测和过滤后的高质量SNP可用于计算种群间的距离。运用Treebest- 1.9.2软件计算距离矩阵,然后通过邻接法(neighbor- joining method)构建系统进化树判断样本间的亲缘关系,使用Bootstrap为置信度检验统计方法,重复抽样次数设为1 000次。运用GCTA软件进行群体主成分分析。利用ADMIXTURE软件进行群体遗传结构分析[26]。群体进化树分析、主成分分析和群体遗传结构分析是群体遗传学的常用分析方法。其中系统进化树是描述群体间分化顺序的分支图或树,可用来揭示群体间的进化关系,并根据群体遗传特征推断它们的亲缘关系[27]。主成分分析是一种研究群体遗传结构的多元统计方法。它主要基于个体基因组中SNP差异的程度,根据不同的性状将个体聚类为不同的亚群,是确定物种亚群数量的有效方法[28]。为了进一步明晰168份种质间的遗传关系,对其遗传结构进行解析。采用PLINK软件进行群体结构分析。首先创建PLINK的输入文件-Ped文件,然后利用ADMIXTURE软件构建群体遗传结构和群体世系信息。
群体遗传多样性 类群的遗传多样性包括核苷酸多样性、观测杂合度(o)、期望杂合度(e)和群体间遗传分化指数(st)。运用vcftools V 0.1.14软件计算单位点核苷酸多样性[()][29–30]。
2 结果和分析
2.1 测序基本数据分析
对168份蓝花楹种质进行了RAD简化基因组测序,共获得测序数据量355.51 Gb,平均2.12 Gb。经过对测序数据的严格过滤,得到高质量的cleandata为353.28 Gb,平均2.10 Gb,平均GC含量为35.14%,碱基质量Q30达到92.10%,表明测序质量较高(表2)。每份种质资源平均获得高质量序列13 920 867条,平均有11 290 873的序列条数可以比对到参考基因组,比对率为81.02%,平均测序深度为23.18×。平均比对率和平均测序深度均满足分析要求,可以进行后续分析。
进行群体SNP的检测,共获得573 704个原始SNP。通过Q20质量控制、SNP的支持数(覆盖深度)在4以上、miss小于20%、maf大于5%过滤和筛选,得到185 054个SNPs;经HWB过滤,共保留60 285个SNP;经LD过滤,共保留45 552个SNP,后续分析均基于HWB和LD过滤后高质量SNP进行分析。
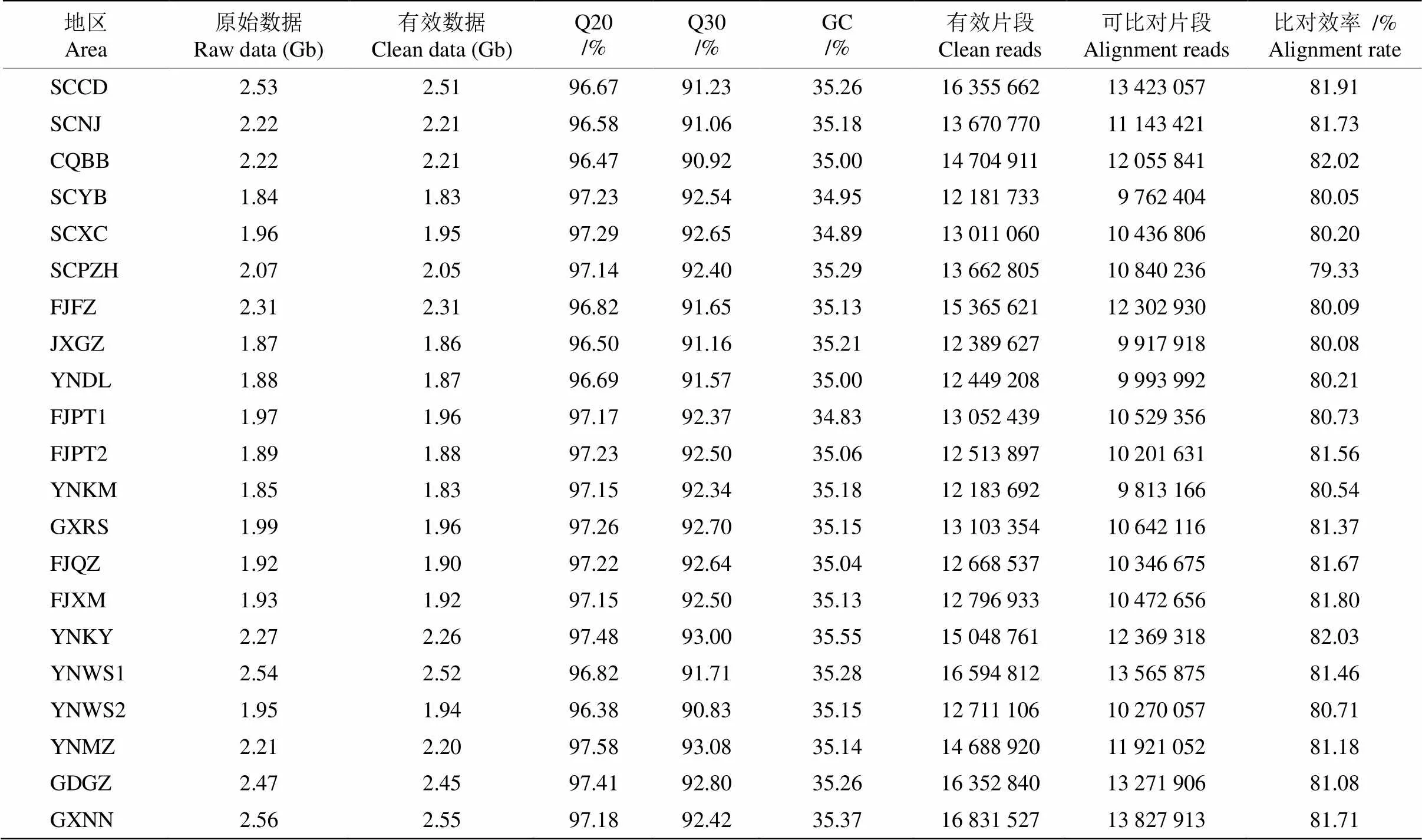
表2 文库构建测序信息
2.2 群体进化树和主成分分析
利用鉴定到的高质量SNP对这168份蓝花楹种质资源构建进化树(图1),可见,供试蓝花楹可分为2大类群,其中SCNJ、CQBB、SCYB、SCXC和SCPZH共5个地区的种质材料归为类群I。其余14个地区的种质材料归为类群II,包括SCCD、YNKM、FJQZ、YNDL、FJPT1、FJPT2、JXGZ、YNKY、GDGZ、GXRS、FJXM、YNMZ、YNWS1、YNWS2、GXNN和FJFZ。从SNP中提取关键信息,采用GCTA软件对168份蓝花楹材料进行PCA主成分分析, 图2为基于3个主成分的PCA聚类图, 可以清楚地反映个体的聚类情况,群体之间的距离能够反映亲缘关系的远近。PCA分析中PCA1、PCA2和PCA3的解释度分别为:0.182 476 058 936 713、0.046 452 359 970 902和0.040 372 436 296 260 5。从3个主成分PC1、PC2和PC3可知:来自SCNJ、SCYB、SCXC、SCPZH、SCCD、CQBB等6个地区的种质材料被归为同一类群;其余13个地区的种质材料归为另一类群, 与群体进化聚类分析结果基本一致。
2.3 群体遗传结构分析
为了确定合适的分群数(K值),用ADMIXTURE软件分析样品的群体结构,预先假定K值为2~15,分别进行运算,并使用交叉验证确定K值,结果表明当K=2时为最优分群,即分为2大类群。结果表明(图3),群体Ⅰ为橙色,种质材料分别来自SCCD、SCNJ、CQBB、SCYB、SCXC和SCPZH等6个地区;群体II为蓝色,种质材料来自其余13个地区。这与群体进化分析、主成分分析结果基本一致。为了解蓝花楹的遗传差异,计算了蓝花楹群体间的遗传分化系数(st)(表3)。可见, 21个蓝花楹群体间的st为0.016~0.178,其中CQBB与FJPT1、CQBB与FJPT2、CQBB与JXGZ的st分别为0.178、0.173和0.174;SCPZH与JXGZ和SCPZH与FJPT1分别为0.175和0.173,st均大于0.15,说明遗传分化较大。FJPT1与FJPT2、YNWS1与YNWS2的st<0.05,说明遗传分化很小,采样时分别对大树和小苗取样的做法无意义。
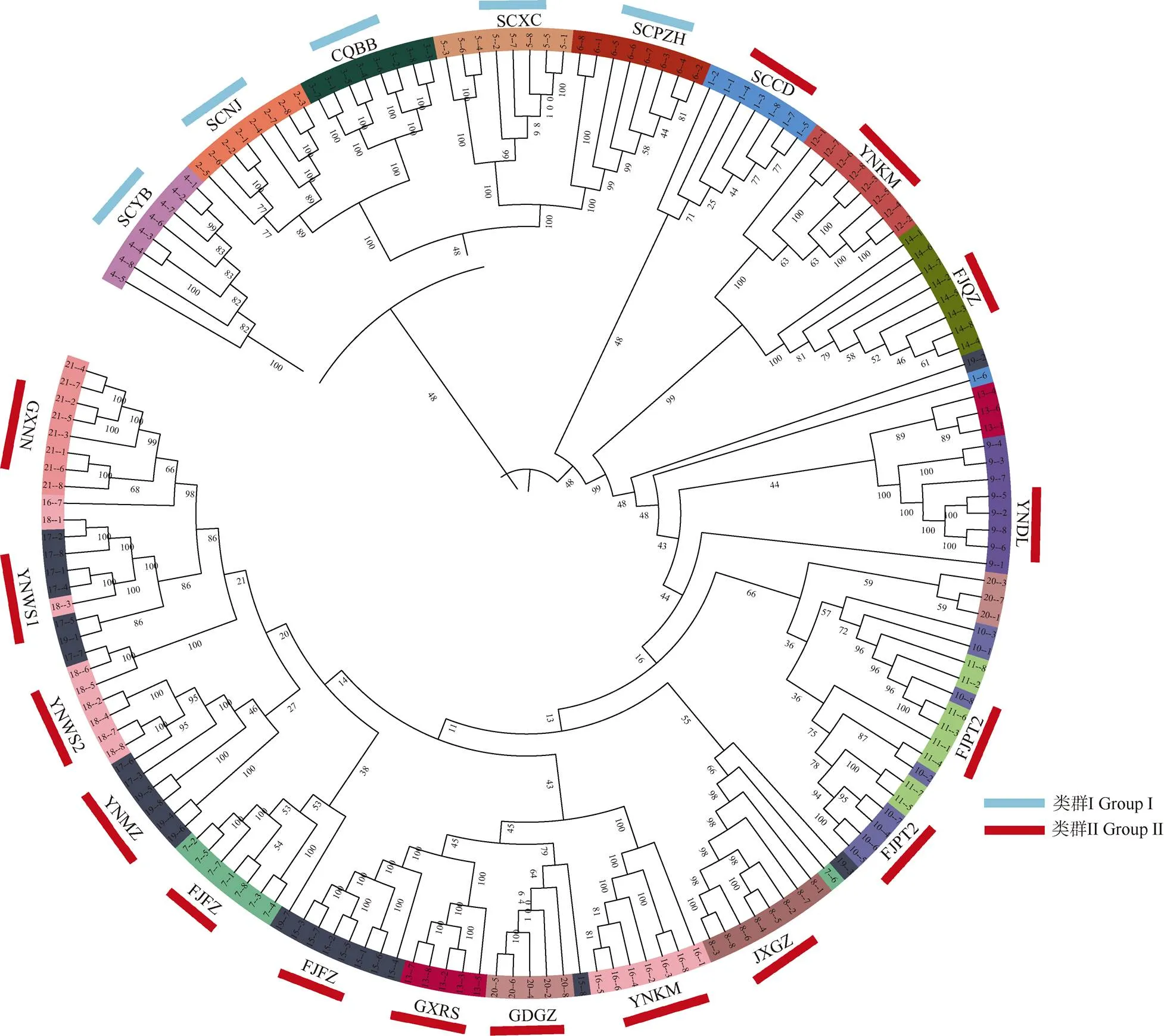
图1 168份蓝花楹资源的群体系统进化树

图2 168份蓝花楹种质资源的群体主成分分析
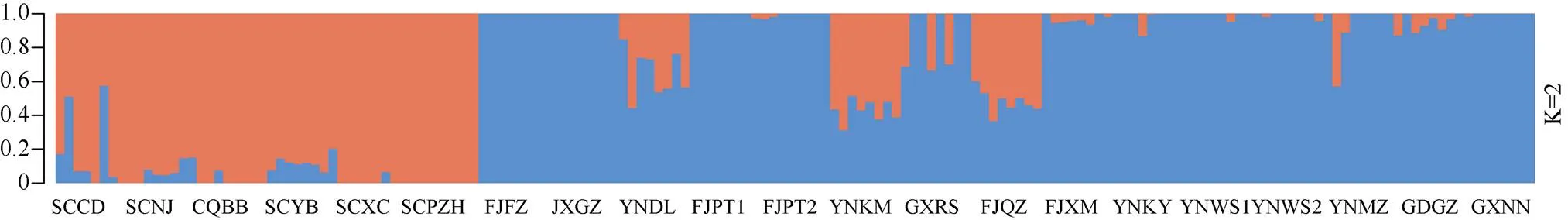
图3 168份蓝花楹种质资源的遗传结构关系
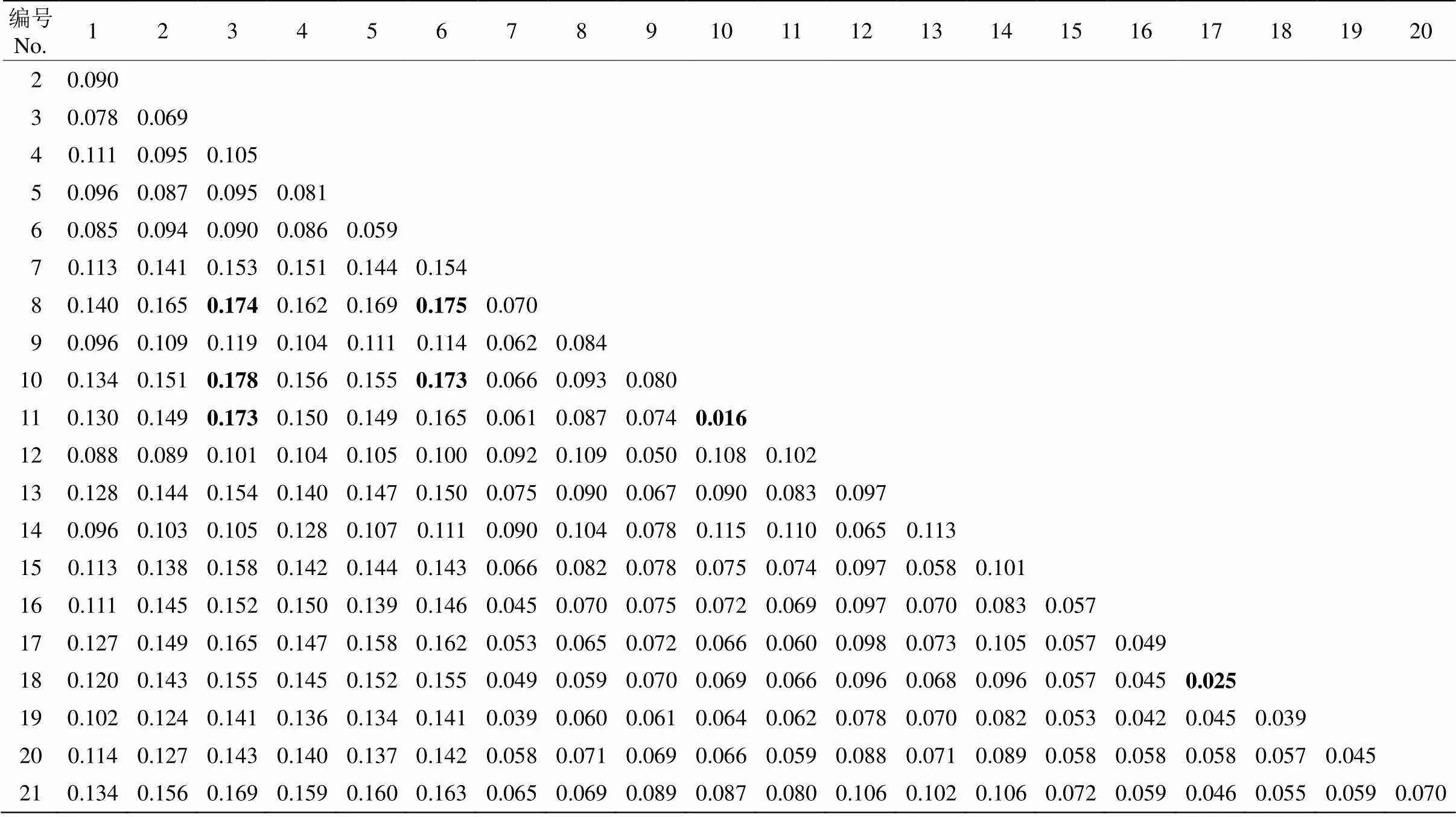
表3 蓝花楹群体间遗传分化系数(Fst)
1~21见表1。
1-21 see Table 1.
2.4 群体遗传多样性分析
为了解蓝花楹群体遗传多样性水平,进行了核苷酸多样性和杂合度分析,核苷酸多样性是指样本中所有可能匹配成对的序列间核苷酸位点差异百分比的平均值,用表示。杂合度反映了群体中的遗传变异程度,杂合度越高,表明群体内遗传多样性越高,包括观测杂合度()和期望杂合度()。从表4可知,居群水平上,蓝花楹的为0.293~0.347, 平均为0.320,为0.406~0.474,平均为0.433,为0.273~0.323,平均为0.298。其中, YNKM居群的和最大,分别为0.347和0.323,表现出最高的遗传多样性;其次为YNDL,、和分别为0.345、0.474和0.321;遗传多样性最小的是GXRS,、和分别为0.293、0.406和0.273。
3 结论和讨论
蓝花楹为国外引进的具有高景观价值的观花乔木,其种质资源利用还处在起步阶段,进行遗传多样性分析和背景研究是十分必要的。本研究首次通过RAD-seq测序技术在全基因组水平上分析蓝花楹群体的遗传结构和遗传多样性,共获得测序数据355.51 Gb,过滤后的高质量clean data为353.28 Gb。测序质量较高, 平均GC含量为35.14%,碱基质量Q20达到97.02%,Q30达到92.10%,说明建库测序成功。共检测获得原始SNP 573 704个,鉴定出185 054个高质量SNP标记,经HWB和LD过滤保留45 552个SNP。这些标记信息将为蓝花楹的遗传育种、保护研究提供了基础。
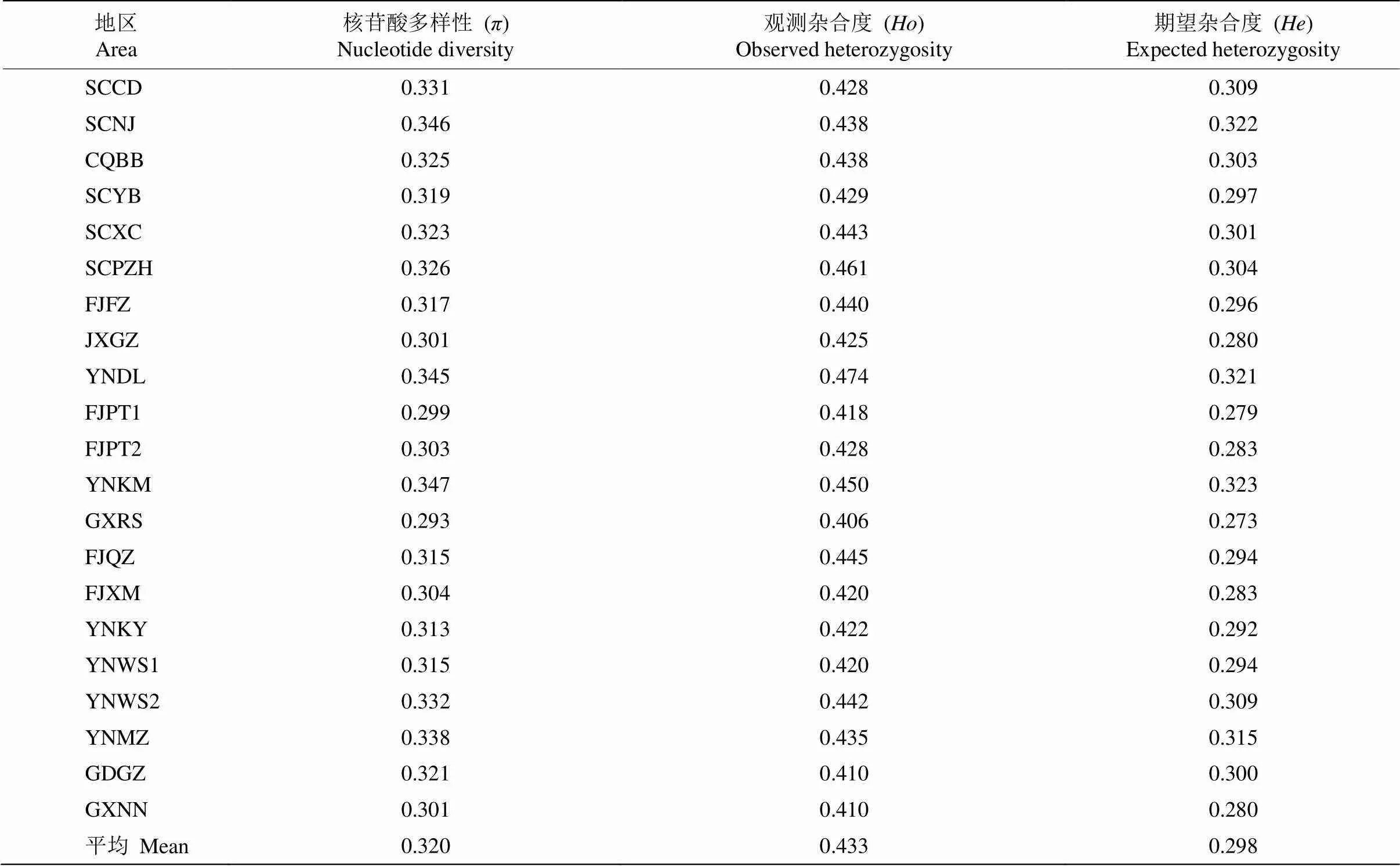
表4 21个蓝花楹群体的遗传多样性参数
对168份蓝花楹种质进行群体进化树构建、主成分分析和群体结构分析,其结果基本一致。群体I的种质材料基本来自川、渝的6个地区,包括SCNJ、SCYB、SCXC、SCPZH、SCCD和CQBB,显示亲缘关系较近,这些地区的行政权属早期均归属四川省管辖,引种栽培时有条件共享同一批种质材料, 说明行政权属归属或地理区域近的材料遗传距离比较接近,这与屈洋等[31]的研究结果一致。由系统进化树的支持率可知,SCNJ、CQBB、SCYB、SCXC和SCPZH的支持率为48%,YNDL和GXRS为44%, YNKY、GDGZ、GXRS和FJXM为43%,YNMZ、YNWS1、YNWS2、GXNN和FJFZ为14%,这些种质无法通过系统进化树分开,可能与种源引入我国后是以种子实生苗繁殖,多年的混乱杂交造成的; 也可能是在来源地已经有充分的基因流,没有形成显著分化。GXRS和YNDL、FJXM,FJFZ和JXGZ以及GDGZ和FJPT群体间呈现出少数的个体混杂现象,表明群体间存在基因交流。
遗传多样性分析基于SNP位点的核苷酸多样性()、观测杂合度()、期望杂合度()[32]。本研究结果表明,21个蓝花楹群体的、和均较高,意味着蓝花楹在SNP水平上的遗传多样性较高, 与海南风吹楠()[33]的研究结果相似。
st是分析群体遗传分化程度的重要指标之一[30],Wright[34]提出,当st为0~0.05时群体间遗传分化很小,可以忽略不计;当st为0.05~0.15时群体间的遗传分化表现中等;当st为0.15~0.25时群体间遗传分化较大;当st大于0.25时,群体间有很大的遗传分化。本研究中群体间的st为0.016~0.178,表明各群体间的遗传分化程度较高。其中,来自川、渝地区的群体I (SCNJ、SCYB、SCXC、SCPZH、SCCD和CQBB)与群体II (FJFZ、JXGZ、YNDL、FJPT1、FJPT2、YNKM、GXRS、FJQZ、FJXM、YNKY、YNWS1、YNWS2、YNMZ、GDGZ和GXNN)的遗传分化程度较高,这与群体系统进化树、主成分分析与群体遗传结构分析结果一致。即两大群体间遗传分化比较大,群体内部居群间遗传分化较小。FJPT1与FJPT2居群和YNWS1与YNWS2居群间的遗传分化最小,说明同一地域大树和小苗采样对遗传多样性和遗传分化无差异,说明该地区蓝花楹种苗无迁移。
尽管蓝花楹最早于20世纪20年代就引入中国,主要作为城市景观树种,但早期保存下来的数量并不多,90年代后期蓝花楹在中国得到了较快推广,遗憾的是,早期引种的蓝花楹的来源地、具体栽培时间等遗传背景均不清晰,后期栽培的蓝花楹基本来自国内种子繁殖培育,或许正是由于这种不清晰的遗传背景,从而难于寻找出类群II中来自其余13个地区种质材料彼此间亲缘关系的远近规律。少数几个种质材料在不同的研究中被划分到不同类群或亚群,呈现出较细微差别,这可能是计算依据不同而导致的,这与王小柯等[35]的研究结果一致。
尽管蓝花楹引种栽培到中国的遗传背景不清,影响了对本研究部分结果的深入研判,但本研究通过对蓝花楹的遗传多样性和群体结构分析,构建起了蓝花楹的遗传图谱,为蓝花楹育种、系统发育研究、遗传保护、分子标记开发以及产业开发应用等提供了理论基础。同时较系统完整的保存了一批早期引种到中国的蓝花楹遗传材料,为后续的深入研究奠定了基础。对于蓝花楹种质资源遗传背景的判定,相比于性状鉴别法和ISSR分子标记[36]来说, RAD-seq技术更具优势,更能准确地反映种质资源间的亲缘关系。
[1] Delectis Florae Reipublicae Popularis Sinicae, Agendae Academiae Sinicae Editta. Florae Reipublicae Popularis Sinicae, Tomus 69 [M]. Beijing: Science Press, 1981.
中国植物志编辑委员会. 中国植物志, 第69卷[M]. 北京: 科学出版社, 1981.
[2] LIU X F, LI X M, ZHANG P Jet alResearch progress ofand the development prospects [J]. Eucalypt Sci Technol, 2015, 32(4): 51–55. doi: 10.13987/j.cnki.askj.2015.04.012.
刘学锋, 李小梅, 张沛健, 等. 蓝花楹的研究进展与开发利用前景 [J].桉树科技, 2015, 32(4): 51–55. doi: 10.13987/j.cnki.askj.2015.04.012.
[3] LI Q. Study on distribution and landscape application ofD. Don in Sichuan region [D]Chengdu: Sichuan Agricul- tural University, 2015.
李青. 蓝花楹在四川地区的分布及园林应用研究[D]. 成都: 四川农业大学, 2015.
[4] FERREIRA N C D F, ROCHA E C, RODRIGUES F, et al.spp. in growth promotion ofD. Don [J]. J Agric Stud, 2021, 9(2): 335–346. doi: 10.5296/jas.v9i2.18410.
[5] NAZ R, ROBERTS T H, BANO A, et al. GC-MS analysis, antimicro- bial, antioxidant, antilipoxygenase and cytotoxic activities ofmethanol leaf extracts and fractions [J]. PLoS One, 2020, 15(7): e0236319. doi: 10.1371/journal.pone.0236319.
[6] GEORGIN J, SALOMÓN Y L D O, FRANCO D S P, et al. Develop- ment of highly porous activated carbon fromseed pods for remarkable removal of aqueous-phase ketoprofen [J]. J Environ Chem Eng, 2021, 9(4): 105676. doi: 10.1016/j.jece.2021.105676.
[7] FARIAS C P, ALVES G S, OLIVEIRA D C, et al. A consortium of fungal isolates and biochar improved the phytoremediation potential ofD. Don and reduced copper, manganese, and zinc leaching [J]. J Soils Sediments, 2020, 20(1): 260–271. doi: 10. 1007/s11368-019-02414-3.
[8] WANG M C, ZHANG L, WANG Z Q. Chromosomal-level reference genome of the neotropical treeD. Don[J]. Genome Biol Evol, 2021, 13(6): evab094. doi: 10.1093/gbe/evab094.
[9] CORDEIRO J M P, LIMA S A A, PAZ S N, et al. Karyotype evolution in the genusJuss. (Jacarandeae, Bignoniaceae): Chromo- some numbers and heterochromatin [J]. Genet Mol Res, 2016, 15(4): gmr15048973. doi: 10.4238/gmr15048973.
[10] ZHAO K K, CHEN R L, WANG J H, et al. Complete plastome sequence ofD. Don (Bignoniaceae): A beau- tiful landscaping tree species [J]. Mitochondr DNA B Resour, 2019, 4(2): 4111–4112. doi: 10.1080/23802359.2019.1692702.
[11] ESCANDÓN A, DE LA TORRE M P, ACEVEDO A, et al. Anchored ISSR as molecular marker to characterize accessions ofL. Don [J]. Acta Hort, 2005, 683: 121–127. doi: 10.17660/ ActaHortic.2005.683.11.
[12] LIU X F, LIU G, LI X M, et al. Analysis of genetic diversity and relationship ofbased on ISSR molecular markers technique [J]. Mol Plant Breed, 2021, 19(21): 7146–7153. doi: 10.13271/j.mpb.019.007146.
刘学锋, 刘果, 李小梅, 等. 基于ISSR分子标记技术的蓝花楹遗传多样性和亲缘关系分析 [J]. 分子植物育种, 2021, 19(21): 7146– 7153. doi: 10.13271/j.mpb.019.007146.
[13] WANG Y K, HU Y, ZHANG T Z. Current status and perspective of RAD-seq in genomic research [J]. Hereditas, 2014, 36(1): 41–49. doi: 10.3724/SP.J.1005.2014.0050.
王洋坤, 胡艳, 张天真. RAD-seq技术在基因组研究中的现状及展望[J]. 遗传, 2014, 36(1): 41–49. doi: 10.3724/SP.J.1005.2014.0050.
[14] ZHANG Y, ZHOU W Y, SUN W. The two major technologies of sequencing based on simplified genome by restriction enzyme dige- stion [J]. Mol Plant Breed, 2020, 18(11): 3562–3570. doi: 10.13271/j. mpb.018.003562.
张羽, 周婉莹, 孙旺. 基于限制性内切酶简化基因组测序的两种主要技术 [J]. 分子植物育种, 2020, 18(11): 3562–3570. doi: 10.13271/j. mpb.018.003562.
[15] XIONG Y, ZHANG J Z, DONG J, et al.A review of reduced- representation genome sequencing technique and its applications in ornamental plants [J]. Acta Hort Sin, 2020, 47(6): 1194–1202. doi: 10.16420/j.issn.0513-353x.2019-0716.
熊燕, 张金柱, 董婕, 等. 简化基因组测序技术在观赏植物中的应用研究进展 [J]. 园艺学报, 2020, 47(6): 1194–1202. doi: 10.16420/j. issn.0513-353x.2019-0716.
[16] ZHAI Z X. SNP mapping and population genetic analyses for 13 Chinese indigenous chicken breeds using red sequencing [D]. Shanghai: Shanghai Jiao Tong University, 2014.
翟正晓. 基于RAD简化基因组测序技术的13种中国地方优良鸡品种SNPs多态性图谱构建及群体遗传学分析 [D]. 上海: 上海交通大学, 2014.
[17] PREMARATHNE M D G P, FUKUTOME N, YAMASAKI K, et al. Elucidation of Japanese pepper (De Candolle) domestication using RAD-Seq [J]. Sci Rep, 2021, 11: 6464. doi: 10. 1038/s41598-021-85909-9.
[18] FENG J Y, ZHAO S, LI M, et al. Genome-wide genetic diversity detection and population structure analysis in sweetpotato () using RAD-seq [J]. Genomics, 2020, 112(2): 1978–1987. doi: 10.1016/j.ygeno.2019.11.010.
[19] FUKUDA S, NAGANO Y, MATSUGUMA K, et al.Construction of a high-density linkage map for bronze loquat using RAD-Seq [J]. Sci Hort, 2019, 251: 59–64. doi: 10.1016/j.scienta.2019.02.065.
[20] HUANG C L, YAO G, TIAN X L, et al. Phylogenomic analysis ofspecies in Guizhou BailiReserve Based on RAD sequencing [J]. Sci Silv Sin, 2021, 57(2): 72–81. doi: 10. 11707/j.1001-7488.20210208.
黄承玲, 姚刚, 田晓玲, 等. 基于RAD高通量测序的贵州百里杜鹃保护区杜鹃花属分类 [J]. 林业科学, 2021, 57(2): 72–81. doi: 10. 11707/j.1001-7488.20210208.
[21] ZHANG S D, YUAN D Y, LU H F, et al. The results of rice germplasm EDV test by genomic analysis and related discussions [J]. Sci Sin Vitae, 2020, 50(6): 633–649. doi: 10.1360/SSV-2020-0068.
张上都, 袁定阳, 路洪凤, 等. 基因组学方法用于水稻种质资源实质派生的检测结果和应用讨论 [J]. 中国科学生命科学, 2020, 50 (6): 633–649. doi: 10.1360/SSV-2020-0068.
[22] ZHANG B D, XUE D X, WANG J, et al. Development and preliminary evaluation of a genomewide single nucleotide polymorphisms resource generated by RAD-seq for the small yellow croaker () [J]. Mol Ecol Resour, 2016, 16(3): 755–768. doi: 10.1111/ 1755-0998.12476.
[23] LI H, DURBIN R. Fast and accurate long-read alignment with Burrows- Wheeler transform [J]. Bioinformatics, 2010, 26(5): 589–595. doi: 10. 1093/bioinformatics/btp698.
[24] LI H, HANDSAKER B, WYSOKER A, et al. The sequence alignment/ map format and SAMtools [J]. Bioinformatics, 2009, 25(16): 2078– 2079. doi: 10.1093/bioinformatics/btp352.
[25] GUI L L. Study on phylogeny ofclade in the genus ofdistributed in China based on RAD-seq [D]. Wuhan: Huazhong Agricultural University, 2017.
桂柳柳. 基于RAD-seq技术的中国葡萄属葛藟葡萄支系的系统发育研究[D]. 武汉: 华中农业大学, 2017.
[26] ALEXANDER D H, NOVEMBRE J, LANGE K. Fast model-based estimation of ancestry in unrelated individuals [J]. Genome Res, 2009, 19(9): 1655–1664. doi: 10.1101/gr.094052.109.
[27] WANG X, GAO M, WU L W, et al. A study on population genetic diversity among(Hemsl.) Gamble in Mount Emei Area [J]. J Plant Genet Resour, 2019, 20(2): 359–369. doi: 10.13430/j. cnki.jpgr.20180812001.
王雪, 高暝, 吴立文, 等. 峨眉山地区杨叶木姜子群体遗传多样性研究 [J]. 植物遗传资源学报, 2019, 20(2): 359–369. doi: 10.13430/j. cnki.jpgr.20180812001.
[28] LI Y F, LI S M, JIN X, et al. Phylogenomic analysis of85species in China based on RAD sequencing [J]. For Res, 2019, 32(3): 1–8. doi: 10.13275/j.cnki.lykxyj.2019.03.001.
李云飞, 李世明, 金鑫, 等. 基于RAD高通量测序探讨中国85种杜鹃花属植物的分类[J]. 林业科学研究, 2019, 32(3): 1–8. doi: 10. 13275/j.cnki.lykxyj.2019.03.001.
[29] YAN S Y, ZHU P, GONG W, et al. Studies on genetic diversity of Juglans cultivar germplasms in Sichuan based on RAD-SNPs analysis [J]. J Trop Subtrop Bot, 2019, 27(1): 19–28. doi: 10.11926/jtsb.3906.
闫思宇, 朱鹏, 龚伟, 等. 基于RAD-SNPs分析的四川核桃良种资源的遗传多样性研究 [J]. 热带亚热带植物学报, 2019, 27(1): 19–28. doi: 10.11926/jtsb.3906.
[30] CAO Y Y, DIAO Q N, CHEN Y Y, et al. Analysis of genetic diversity of melon based on 2b-RAD simplified genome sequencing [J]. Acta Bot Boreali-Occid Sin, 2021, 41(1): 96–106. doi: 10.7606/j.issn.1000- 4025.2021.01.0096.
曹燕燕, 刁倩楠, 陈幼源, 等. 基于2b-RAD简化基因组测序的甜瓜遗传多样性分析 [J]. 西北植物学报, 2021, 41(1): 96–106. doi: 10. 7606/j.issn.1000-4025.2021.01.0096.
[31] QU Y, ZHOU Y, WANG Z, et al. Analysis of genetic diversity and structure of tartary buckwheat resources from production regions [J]. Sci Agric Sin, 2016, 49(11): 2049–2062. doi: 10.3864/j.issn.0578-1752. 2016.11.002.
屈洋, 周瑜, 王钊, 等. 苦荞产区种质资源遗传多样性和遗传结构分析 [J]. 中国农业科学, 2016, 49(11): 2049–2062. doi: 10.3864/j. issn.0578-1752.2016.11.002.
[32] ZHANG S S, KANG H M, YANG W Z. Population genetic analysis ofby reduced-representation sequencing technique [J]. Bull Bot Res, 2019, 39(6): 899–907. doi: 10.7525/j.issn.1673-5102. 2019.06.013.
张珊珊, 康洪梅, 杨文忠. 基于简化基因组技术的云南蓝果树群体遗传分析 [J]. 植物研究, 2019, 39(6): 899–907. doi: 10.7525/j.issn. 1673-5102.2019.06.013.
[33] CAI C N, HOU Q X, CI X Q, et al. Genetic diversity of: An endangered species with extremely small populations [J]. J Trop Subtrop Bot, 2021, 29(5): 547–555. doi: 10.11926/jtsb.4364.
蔡超男, 侯勤曦, 慈秀芹, 等. 极小种群野生植物海南风吹楠的遗传多样性研究 [J]. 热带亚热带植物学报, 2021, 29(5): 547–555. doi: 10.11926/jtsb.4364.
[34] WRIGHT S. The interpretation of population structure by F-statistics with special regard to systems of mating [J]. Evolution, 1965, 19(3): 395–420. doi: 10.2307/2406450.
[35] WANG X K, JIANG D, SUN Z Z. Study on phylogeny of 240 mandarin accessions with genotyping-by-sequencing technology [J]. Sci Agric Sin, 2017, 50(9): 1666–1673. doi: 10.3864/j.issn.0578-1752.2017.09.012.
王小柯, 江东, 孙珍珠. 利用GBS技术研究240份宽皮柑橘的系统演化[J]. 中国农业科学, 2017, 50(9): 1666–1673. doi: 10.3864/j.issn. 0578-1752.2017.09.012.
[36] YANG Y L, MA X Q, ZHANG M Q. ISSR molecular marker and its application to the research on genetic breeding of trees [J]. Subtrop Agric Res, 2006, 2(1): 18–24. doi: 10.3969/j.issn.1673-0925.2006.01.006.
杨玉玲, 马祥庆, 张木清. ISSR分子标记及其在树木遗传育种研究中的应用 [J]. 亚热带农业研究, 2006, 2(1): 18–24. doi: 10.3969/j. issn.1673-0925.2006.01.006.
Population Genetic Analysis ofby RAD Hight Throughput Sequencing Technique
LIU Xuefeng1, LI Xiaomei2, ZHANG Guowu1*, ZHANG Peijian1, LIU Guo1, HUANG Min1, CHEN Yinxia3
(1. China Eucalypt Research Centre,Zhanjiang 524022, Guangdong, China; 2. Zhanjiang University of Science and Technology,Zhanjiang 524000, Guangdong,China; 3.Jiangxi Forestry Sci-tech Promotion and Propaganda Education Center,Nanchang 330033, China)
In order to understand the genetic diversity ofgermplasm resources, 168 germplasms ofwere restriction-site associated DNA sequencing (RAD-seq), and then the phylogenetic tree was constructed, principal component (PCA), population structure and genetic diversity were analyzed. The results showed that the mean alignment rates to the reference genomes were 81.02% with an average sequencing depth of 23.18×. After cleaned and filtered, a total of 45 552 SNPs were obtained. All tested germplasms could be clustered into two groups, those from Sichuan-Chongqing region were in one group, and the rest were in another group.has a high genetic diversity at the SNP level. Among them, nucleotide diversity (π) and expected heterozygosity () in YNKM population is the largest, showing the highest genetic diversity. Therefore,from Sichuan-Chongqing region showed relatively close genetic relationship, suggesting that they might from the same ancestor, and those from other regions might be randomly introduced and cultivated.
; RAD-seq; Genetic diversity; Population genetic
10.11926/jtsb.4517
2021-09-02
2021-12-13
中央级公益科研院所专项资金项目(CAFYBB2019MB004);广东省林业科技创新项目(2018KJCX024)资助
This work was supported by the Project for Central Public Research Institutes (Grant No. CAFYBB2019MB004), and the Project for Forestry Science and Technology Innovation in Guangdong (Grant No. 2018KJCX024).
刘学锋(1985生),工程师, 从事森林培育及园林植物学研究。Email: cercliuxf@caf.ac.cn
. E-mail: fyzgwu@163.com
