2例婴儿CDKL5基因新发突变致早发性癫痫脑病的临床特征与遗传学分析
2022-10-15杨少青刘鸿丽宋婷婷姜永生张小鸽
董 玲,谢 云,杨少青,刘鸿丽,王 琪,宋婷婷,姜永生,张小鸽
(1.西北妇女儿童医院a.检验科;b.神经内科,西安 710061;2.军事口腔医学国家重点实验室,口腔疾病国家临床医学研究中心,陕西省口腔医学重点实验室/空军军医大学口腔医学院口腔罕见病与遗传病门诊/口腔生物学教研室,西安 710032;3. 西安交通大学第二附属医院检验科,西安 710004)
细胞周期蛋白依赖性蛋白激酶5(cyclin-dependent kinase-like 5,CDKL5)缺乏症(CDD)是一种严重的神经发育性疾病,由位于X染色体的CDKL5(OMIM 300203)基因突变导致[1]。大多数CDKL5基因变异的个体表现出相似表型,包括早发难治性癫痫[2]、严重的全身性发育迟缓和运动功能明显受损[3]。CDKL5基因变异致癫痫通常开始于出生后的前三个月,表现为对抗癫痫药物治疗反应不佳的各种癫痫发作[4]。由于治疗手段有限,CDD患者可能会经历癫痫性脑病的永久性症状和显著的发育障碍[5]。本研究回顾性分析2例CDKL5基因新发突变致患儿早发性癫痫脑病的临床特征及基因变异特点。
1 材料与方法
1.1 研究对象 病例1,女,1月17天,因反复抽搐发作26天入院。患儿出生后20天无明显诱因出现抽搐发作,表现为双手握拳、肢体强直,持续时间约10s后可自行缓解,发作1~2次/天。出生后24天患儿抽搐较前频繁,多达10余次/天,表现为:双眼向上凝视、眨眼、口唇发绀、牙关紧闭、双手握拳、口唇及四肢抖动,持续时间15~40s。患儿系第1胎第1产,足月剖宫产,出生体重4.04kg,无病理性黄疸,家族史无异常。体格检查:发育正常、神志清楚、精神尚可、皮肤及面容未见异常、心肺腹无异常、四肢肌张力正常、病理征阴性。实验室检查:血细胞检测、尿常规、生化(肝功、肾功、心肌酶、电解质、血氨、铜蓝蛋白)、甲状腺功能等检查均无异常。染色体核型无异常。头颅核磁共振成像(MRI)示双侧颞前极脑外间隙增宽。长程视频脑电监测显示发作间期可见中央、顶导为著较多量中-高波幅尖波、棘波、尖棘慢复合波、慢波及活动发放,两侧不同步。发作期有两种表现形式:第一种形式为全面强直阵挛发作,第二种发作形式为不典型痉挛发作。患儿采用左乙拉西坦与苯巴比妥联合治疗,治疗后发作次数明显减少。
病例2,女,1月26天,因发作性抽搐20天入院。患儿出生后1月睡眠过程中出现一次抽搐,表现为双眼凝视、双上肢上抬、双下肢屈曲抖动,持续约10s后缓解。其后一周患儿抽搐发作频繁,最多7~8次/天,每次持续10~20s。患儿系第1胎第1产,足月剖宫产,出生体重3.05kg,无出生缺氧窒息史、家族史无异常。体格检查:发育正常、神志清楚、精神反应一般、头颅无畸形,面容、皮肤未见异常,心肺腹无异常,四肢肌张力正常,病理反射(-)。实验室检查:患儿谷氨酸氨基转移酶(ALT)为69.9 U/L,天冬氨酸氨基转移酶(AST)为70.6 U/L,其余血细胞检测、尿常规、生化(肾功、心肌酶、电解质、血氨、无机磷、铜蓝蛋白)、甲状腺功能等检查均无异常。染色体核型无异常。MRI无异常。长程视频脑电监测显示发作间期清醒和睡眠期可见较多量多灶性中-高波幅尖波、棘波、尖棘慢复合波及活动发放,发作期表现为全面性强直阵挛发作。患儿采用托吡酯、苯巴比妥联合治疗,治疗效果欠佳,后患儿家属要求出院。
1.2 仪 器 与 试 剂 BloodGen Midi Kit基 因 组 提取试剂盒(北京康为世纪生物科技有限公司);NimbleGen序列捕获实验建库试剂盒(罗氏诊断公 司);Illumina hiseq2500第 二 代 测 序 仪(美 国Illumina公司);ABI 3730XL 测序仪(美国应用生物系统公司(Applied Biosystems)。
1.3 方法
1.3.1 基因组DNA提取:研究通过西北妇女儿童医院伦理委员会审批,经患儿家长知情同意后,抽取患儿与其父母外周血,采用血液DNA提取试剂盒,按照试剂盒说明书步骤提取血液基因组DNA。
1.3.2 目标基因捕获和全外显子组测序:参考相关文献与OMIM数据库信息,将与癫痫相关遗传病基因的基因组外显子区域应用罗氏NimbleGen捕获探针进行目标基因全外显子捕获。采用Illumina hiseq2500平台进行测序,平均测序深度100×~200×,测序得到图像原始数据,采用basecall分析软件BclToFastq得到原始数据。应用BWA软件将测序数据与参考基因组(hg19基因组)序列进行比对统计。
1.3.3 数据筛选及生物信息学分析:分别采用分析软件samtools和pindel进行SNP及Indel检测及注释。根据测序深度、突变质量对检测得到的SNP,Indel进行过滤筛选,得到高质量可靠的突变。使用人类基因组变异软件Provean,SIFT2等软件基于同源比对,蛋白结构的保守性等的算法,预测筛选出的变异对蛋白质的影响。对剪切位点附近的突变,做剪切危害性预测。筛选千人基因组(1 000 Genomes)、ExAC数据库、ESP6500数据库中突变频率<0.001且预测结果为致病性的位点。采用美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics ,ACMG)遗 传变异分类标准与指南评价突变致病性。
1.3.4 Sanger测序验证:根据CDKL5基因验证位点序列设计引物,采用PCR方法进行扩增。PCR反应条件为:95℃预变性5 min;95℃变性30 s,60℃退火30 s,72℃链延伸30s,扩增30个循环;最后72℃补充延伸10 min。PCR 的体系均为50μl。例1测序引物序列为:正向:5'-TCCACTGCACACC AAAACCT-3',反向:5'-GAGGACGTTGCTGGGA AGAA-3'。例2测序引物序列为:正向:5'-CCGC TCAGAACCATGACTGT-3',反向:5'-ACTTATTTG TGGGAGACTGGGT-3'。
2 结果
外显子测序及Sanger测序结果显示例1患儿X染色体 上CDKL5基 因 存在c. 1307C>G(p.Ser436*)无义突变,导致蛋白质436位提前终止,见图1。例2患儿X染色体上CDKL5基因存在c.1974_1975del(p.Val659GlyfsTer23)杂合移码突变,见图2。目前尚无数据库收录上述突变。根据ACMG遗传变异分类标准与指南,例1与例2患儿突变致病证据均为PVS1+PS2,综合判断属于致病突变(pathogenic variants)。

图1 例1家系CDKL5突变位点Sanger测序图

图2 例2家系CDKL5突变位点Sanger测序图
本研究2例患儿的临床特点汇总如下:①癫痫发作均在出生后1个月以内(最大30 天);②发作形式均以部分运动性发作起病。例2患儿随访一年,目前联用3种抗癫痫药物(托吡酯,氨己烯酸片,吡仑帕奈片)治疗一年,加生酮饮食治疗8个月,患儿目前仍有癫痫发作,2~3天发作一次,发作形式无变化。患儿目前一岁,全面性发育落后,竖头稳,不会坐,翻身欠佳,偶可逗笑,反应慢。例1患儿随访2个月,因苯巴比妥导致睡眠增多的副作用,更换抗癫痫药为奥卡西平+左乙拉西坦治疗,目前控制较好,偶有癫痫发作。因其年龄小,目前尚不清楚对运动智力发育的影响,需继续随访。
3 讨论
细胞周期蛋白依赖性蛋白激酶5(CDKL5)蛋白属于类周期蛋白依赖性激酶家族成员,该家族共有5个成员,即CDKL1~CDKL5。根据ProteomicsDB数据库汇总信息,该家族成员中只有CDKL2和CDKL5在大脑中表达,其中CDKL5在大脑皮质表达最高,见图3。表明CDKL5基因突变对大脑皮质功能可能造成较大影响[6]。多篇研究也确认CDKL5在小鼠大脑皮层、海马、丘脑、纹状体和嗅球中表达最高,表明CDKL5可能参与认知能力与随意运动的调控[7]。
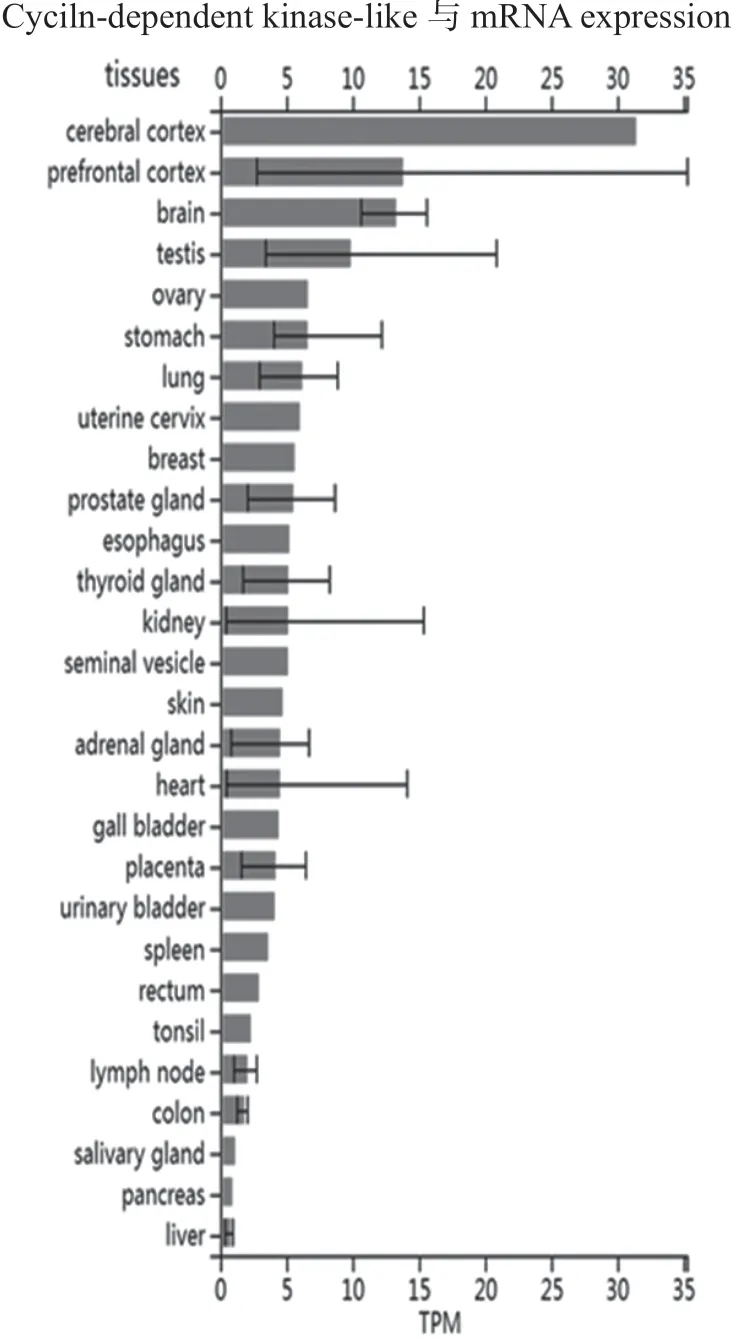
图3 CDKL5基因转录表达谱
CDKL5蛋白是分子量约为116 KDa的大分子量蛋白。主要分为N端和C端两个区域,N端包括了ATP结合区与丝/苏氨酸蛋白激酶活性位点,决定了CDKL5的激酶功能;C端约包括700个氨基酸,它似乎是激酶活性的调节区域,并且具有核定位序列[8]。2003年首次发现CDKL5突变会导致X连锁显性遗传的CDD[9]。后续临床研究发现CDD患者多为女性,临床症状较为多样,包括早发性癫痫、智力发育迟缓、肌张力减退、小头畸形、语言功能障碍、易激动、自闭症等,部分患者症状与Rett综合征以及Angelman综合征非常类似[10]。CDKL5参与人体磷酸化信号通路,一些致病性CDKL5变异体表现出磷酸化活性降低或缺失,导致严重的早发性神经发育疾病[11]。已有研究表明,上游信号使CDKL5在细胞核和细胞质之间穿梭,且定位依赖于细胞周期[12]。因此,CDKL5定位是不断变化的,这些变化使CDKL5能够磷酸化细胞各部分中的多种蛋白质。由于CDD与甲基化CpG结合蛋白2(MECP2)突变导致的Rett综合征症状非常类似,研究者长期以来推测CDKL5介导MECP2磷酸化,但后续实验表明CDKL5催化MECP2磷酸化的活性并不强[13]。有文献表明,CDKL5可能是通过催化DNA甲基转移酶1(DNMT1),以及组蛋白去乙酰化酶4(HDAC4)的磷酸化,从而间接影响MECP2活性,乃至影响机体神经功能[14-15]。此外,研究表明,CDKL5缺乏会干扰170 kDa(CLIP170)/Rac1三元复合物形成的IQGAP1/细胞质连接蛋白,并对CLIP170的微管结合产生负面影响[16]。敲除CDKL5可阻止锥体生长,抑制轴突延长,并减少极化神经元。神经活性类固醇孕烯醇酮合成衍生物孕烯醇酮甲醚(3β-甲氧基孕烯醇酮)已被确证可通过恢复CLIP170的微管结合修复CDKL5缺陷神经元的形态学缺陷[16-17]。因此,此类药物的开发和使用可能是一种有效的治疗策略。
到目前为止,已有超过265种CDKL5基因的致病性变体被报道。大约50%的变异是点突变,错义突变是最常见的(38%)。然而,只有27%被认为是致病性的,致病性错义突变发生在编码CDKL5催化结构域的区域,导致酶的活性降低,特别是其修饰氨基酸残基的能力大幅度降低[18]。移码突变与无义突变通常会导致CDD,CDKL5 C末端区域包含一个对调节该蛋白在细胞内定位至关重要的序列,CDKL5的无义突变使得缺乏C末端的蛋白质形成,从而造成异常的定位模式,如在体外瞬时表达[19]。此外,过早终止密码子触发CDKL5 mRNA的无义介导衰变(NMD)导致CDKL5的表达缺失也可能引起CDD[20]。CDKL5突变位置与CDD症状具有一定的相关性。目前认为N端突变的患者有更严重的运动障碍,包括难治性癫痫与小头症,而C末端突变的患者症状较轻[21]。
对比两例患儿症状与突变类型可以发现,二者均属于C末端位置出现的功能缺失(lost of funtion)突变,症状均为早发性癫痫,未发现小头畸形等临床症状。但例1患儿癫痫发作频率较低,治疗效果较好;而例2患儿发作频率较高,治疗效果不佳。本研究认为症状的差异与两例先证者CDKL5的突变类型相关。病例1突变导致CDKL5翻译在外显子12中段(c.1307)处提前终止;病例2使CDKL5在外显子13(c.1974)位发生移码突变,导致c.2043位产生提前终止,蛋白产物不仅被截短,且C端23个氨基酸残基与野生型不同,见图4。如前所述,C端对CDKL5蛋白的细胞内定位具有调控作用,因此该突变很可能影响CDKL5蛋白细胞内定位。已有文献报道,CDD患者移码突变与无义突变产生的错误mRNA会受到无义突变导致的RNA降解效应(nonsense-mediated mRNA decay,NMD)影响,从而被细胞识别降解,因此症状较错义突变更轻[22]。病例2的提前终止发生在13号外显子末端,根据NMD降解效应的50边界规则[23],该突变不会触发NMD降解效应,导致突变蛋白被翻译,因此症状更重,治疗效果较差。而病例1的提前终止出现在外显子12中段,距离转录剪接点超过50bp,可以触发NMD效应,导致突变蛋白被降解,因此症状较轻。

图4 两个突变的转录产物分析
综上所述,本研究丰富了早发性癫痫脑病致病基因CDKL5的突变谱,并讨论了突变类型与症状的相关性,为遗传咨询和产前诊断提供了可靠的依据。
