SARS-CoV-2病毒感染潜在关键分子生物标志物及免疫浸润特征分析
2022-10-11于敏王敏魏延焕刘毅毅
于敏,王敏,魏延焕,刘毅毅
1.日照市人民医院急诊医学科,山东 日照276800;
2.海军军医大学第三附属医院检验科,上海200438
新型冠状病毒肺炎(corona virus disease 2019,COVID-19)是一种由严重急性呼吸综合征冠状病毒2(severe acute respiratory syndrome-coronavirus 2,SARS-CoV-2)感染造成的严重传染病[1]。目前,已在世界范围内持续流行时间超过2年,致使人类生命健康及经济发展面临严重威胁[2]。随着对SARS-CoV-2病毒相关研究的不断深入及治疗方法的不断改进,感染患者的治愈率持续攀升,病死率不断降低[3]。研究表明,COVID-19发病率持续增长是由SARS-CoV-2不断突变产生新的高毒毒株引起的,且病毒刺突蛋白的突变与当前疫苗接种的有效性降低有关[4]。SARS-CoV-2毒株发生突变后感染力和病毒载量显著增加[5]。因此,筛选SARSCoV-2病毒与人体免疫细胞相关的关键分子生物标志物对确定免疫治疗的潜在靶标至关重要。
基因表达综合(gene expression omnibus,GEO)数据库是目前最大的基因组测序数据库之一,包含来自COVID-19患者免疫细胞的详细基因组测序信息[6]。加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)是一种广泛使用的生物信息学工具,可用于识别具有高协同变异的基因集[7]。GSE152418数据集[8]是基于SARS-CoV-2病毒感染与机体免疫系统之间相互作用研究较少的背景下开展的,通过对外周血单核细胞的转录组学分析探究COVID-19患者的免疫反应情况。本研究从GEO数据库下载GSE152418数据集,综合运用生物信息学方法筛选SARS-CoV-2病毒感染潜在的关键分子生物标志物并分析其免疫浸润特征,旨在为开发新的免疫治疗的潜在靶标提供依据。
1 材料与方法
1.1 资料来源
从GEO数据库下载GSE152418数据集(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152418),测序平台为GPL24676 Illumina NovaSeq 6000(Homo sapiens)。数据集共包含34例样本,样本来源于外周血单核细胞,其中COVID-19患者17例,健康对照17例。样本数据由美国亚特兰大市耶克斯国家灵长类动物研究中心提供。
1.2 差异表达分析
对数据矩阵中低表达基因进行过滤后,用“DESeq2”包[9]进行差异分析。高表达基因确定标准为log2FC≥1,P<0.05;低表达基因确定标准为log2FC≤-1,P<0.05。保存差 异基因(differential genes,DEGs)用于后续分析,并用热图和火山图进行可视化。
1.3 加权基因共表达网络分析
用“WGCNA”包[10]进行WGCNA分析。①数据进行归一化及对数化处理,选择方差>25%的基因进行分析;②弃除离群样本;③确定最佳邻接函数参数(软阈值),构建邻接矩阵,后转为拓扑重叠矩阵,计算基因间相异度;④使用动态剪切树划分基因模块,合并相关性系数大于0.8(即相异性系数小于0.2)的模块;⑤计算模块与临床特征的相关性。选择与临床特征最相关的模块计算基因显著性(gene significance,GS)和模块隶属度(module membership,MM)。最后,用韦恩图提取模块基因与DEGs的共同基因(common genes,CGs)用于后续分析。
1.4 基因功能及信号通路富集分析
用“ClusterProfiler”包[11]对1.3步 骤 得 到 的CGs进行GO功能及KEGG信号通路富集分析。
1.5 蛋白互作网络分析
利用String数据库(https://string-db.org/)构建蛋白 互 作(protein-protein interaction,PPI)网络,最小交互分数设置为0.9。然后将结果在Cytoscape3.8.2中进行可视化,利用插件CytoHub-ba筛选关键基因(Hub)。
1.6 miRNA-转录因子-mRNA网络分析
分 别 利 用miRTarBase(https://mirtarbase.cuhk.edu.cn/~miRTarBase)、Starbase(https://starbase.sysu.edu.cn/starbase2)和Targetscan(https://www.targetscan.org/vert_72)预测调控Hub基因的miRNA。利用韦恩图取3个数据库预测的共同miRNA,以提高预测的准确性。利用Enrichr(https://maayanlab.cloud/Enrichr)数据库预测调控Hub基因的转录因子(TF)。再用Cytoscape3.8.2构建可视化miRNA-TF-mRNA调控网络。
1.7 免疫浸润特征分析
用CIBERSORT算法[12]分析样本中浸润的免疫细胞构成比例。用“Vioplot”包[13]分析COVID-19与健康对照样本间免疫细胞的水平及差异。用“Corrplot”包[12]绘制相关性热图。
2 结果与分析
2.1 差异表达基因分析结果
DEGs的可视化见图1,共得到2 049个DEGs,其中上调及下调基因分别有1 873个及176个。

图1 差异基因的可视化图Fig.1 Visualization of differential genes
2.2 WGCNA结果
共得到方差大于25%的基因4 743个。设置阈值h=120,剔除2个异常样本,保留聚类1中的32个样本。选择最佳软阈值β=4(R2=0.9)使基因表达关系符合无尺度网络(图2A)。通过动态剪切树划分基因模块,设置每个模块的最低基因数为50,合并相近的模块(即相异性系数小于0.2)(图2B)。共得到7个模块,其中“土耳其蓝色”模块基因与SARS-CoV-2感染这一临床特征相关性最高(r=0.91,P<0.001)(图2C)。“土耳其蓝色”模块中MM和GS之间呈显著正相关(r=0.96,P<0.001)(图2D)。韦恩图结果共得到766个CGs(图2E)。
2.3 CGs功能及信号通路富集分析结果
功能及信号通路富集结果(表1~2)显示,CGs主要参与有丝分裂、微管结合、阳离子通道活性及卵母细胞减数分裂、细胞衰老、心肌病等。

表1 GO功能注释及富集分析Table 1 GO functional annotation and enrichment analysis
2.4 PPI网络分析结果
如图3所示,利用String数据库及Cytoscape3.8.2软件构建CGs的PPI网络图,然后利用CytoHubba插件筛选到Top10的Hub基因分别为CDK1、BUB1、CCNA2、CDC20、KIF11、BUB1B、CDCA8、TOP2A、CCNB2、KIF20A。

图3 CGs的PPI网络图Fig.3 PPI network diagram of CGs
2.5 miRNA-TF-mRNA调控网络分析
通过miRTarBase、Starbase、Targetscan及Enrichr数据库分别预测调控Hub基因的miRNA和TF后,在Cytoscape3.8.2软件中构建miRNA-TFmRNA调控网络,调控网络中包含51个miRNA、5个TF和10个mRNA,其相互作用关系见图4。

图4 Hub基因的miRNA-TF-mRNA调控网络图Fig.4 miRNA-TF-mRNA regulatory network of Hub genes
2.6 免疫细胞浸润分析
根据CIBERSORT算法预测COVID-19患者和健康对照组间的免疫细胞浸润水平。样本中22种免疫细胞的相对百分比见图5。与健康对照组比较,COVID-19患者幼稚B细胞、嗜酸性粒细胞浸润水平显著降低(P<0.05),浆细胞、活化肥大细胞浸润水平显著升高(P<0.05)(图6)。相关性分析结果显示,活化肥大细胞与嗜酸性粒细胞(r=0.83,P<0.05),静息肥大细胞与单核细胞(r=0.77,P<0.05)呈显著正相关。静息自然杀伤细胞与静息肥大细胞(r=-0.69,P<0.05)及单核细胞(r=-0.68,P<0.05)呈显著负相关(图7)。

图5 免疫细胞相对百分比图Fig.5 Relative percentage of immune cells

图6 两组样本间免疫细胞浸润水平比较图Fig.6 Comparison of immune cell infiltration levels between the two groups of samples

图7 免疫浸润细胞亚型间相关性分析图Fig.7 Correlation analysis of immune infiltrating cell subtypes
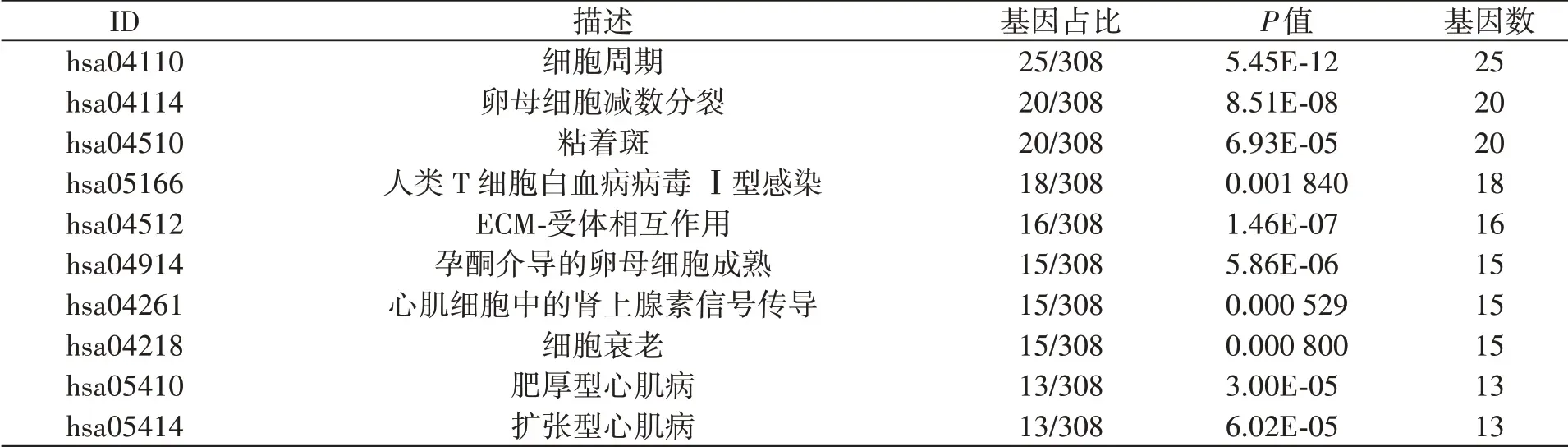
表2 KEGG信号通路富集分析Table 2 KEGG signaling pathway enrichment analysis
3 讨论
COVID-19已在全球多个国家和地区造成大流行。据世界卫生组织(World Health Organization,WHO)官网统计数据显示,截至2022年9月全球确诊COVID-19病例已超过6.03亿例,死亡人数超过648万,但其感染的分子机制仍不完全明确[14]。本研究对包含17例COVID-19患者和17例健康对照样本的高通量测序数据集进行分析,共筛选出2 049个DEGs,其中1 873个基因表达上调,176个基因表达下调。通过WGCNA分析得到7个模块,其中“土耳其蓝色”模块与COVID-19相关性最高。利用韦恩图得到766个CGs。WGCNA分析更侧重于识别整个模块中功能相似的基因,而不是单个基因的差异表达[15]。通过富集分析发现,CGs主要参与有丝分裂、微管结合、阳离子通道活性及卵母细胞减数分裂、细胞衰老、心肌病等,富集结果的差异性也可能解释了COVID-19临床表现和患者预后的异质性[16]。
本研究进一步利用String数据库对CGs进行PPI网络分析,筛选到的前10位Hub基因分别为CDK1、BUB1、CCNA2、CDC20、KIF11、BUB1B、CDCA8、TOP2A、CCNB2、KIF20A。Hahn等[17]研究发现,细胞周期蛋白依赖性激酶1(CDK1)与抑制SARS-CoV-2病毒复制有关。Agrawal等[18]也发现BUB1是抗SARS-CoV-2治疗的潜在靶点。Kim等[19]研究表明,Bcl-2抑制剂ABT-737介导的Bcl-2抑制与细胞周期蛋白A2(CCNA2)和细胞周期蛋白B1(CCNB1)的低表达有关,而Bcl-2抑制剂对SARS-CoV-2病毒的活性有抑制作用。此外,构建的miRNA-TF-mRNA调控网络中调控Hub基因的miRNA 51个、TF 5个。与多数病毒一样,SARSCoV-2病毒RNA与宿主发生相互作用最关键的步骤是通过miRNA靶向调控RNA干扰宿主基因表达[20]。此外,5个TF蛋白是Hub基因的关键转录调节因子,与免疫缺陷及多种癌症有关[21]。
免疫应答在COVID-19发病机制中的关键作用越来越受到关注,包括固有免疫和适应性免疫细胞浸润特征[22]。本研究利用CIBERSORT算法评估了22种免疫细胞在样本中的浸润水平[12]。结果表明,COVID-19患者较健康对照样本幼稚B细胞、嗜酸性粒细胞浸润水平显著降低,浆细胞、活化肥大细胞浸润水平显著升高。研究表明,B细胞的功能状态及分化情况与COVID-19患者的预后有关[23]。Kuri-Cervantes等[24]研究发现,与健康对照及轻中症COVID-19患者比较,重症患者的外周血B细胞水平降低,但浆细胞水平升高,表明SARS-CoV-2病毒感染导致机体B细胞数量减少且分化异常,这可能也是COVID-19病情程度不一的原因之一。Zhang等[25]研究发现,COVID-19住院患者中大多数嗜酸性粒细胞低于正常水平,这有望作为诊断时的参考指标之一。Nagashima等[26]研究发现,COVID-19患者中存在更多的活化肥大细胞,这些细胞与影响患者病情严重程度的血管通透性过高、水肿和弥漫性肺泡损伤事件直接相关。Sun等[27]利用GSE152418数据集及多中心肾移植受者队列数据发现,受COVID-19影响,肾移植受者血液转录组结果显示T细胞和适应性免疫激活显著减少。
本研究也具有一定的局限性:第一,本研究样本数据是从GEO数据库中获取的,数据受到样本量、临床信息缺乏等因素影响,使得研究结论的可靠性受限;第二,对于免疫浸润特征的分析尚不能证实免疫细胞与miRNA、TF、mRNA等标志物之间的相互作用关系。因此,今后应开展体内外试验研究,进一步验证本研究结论的准确性。
综上所述,本研究通过WGCNA及PPI网络分析筛选出10个Hub基因,并预测到调控Hub基因的5个TF及51个miRNA,且COVID-19患者与健康对照的免疫浸润特征存在显著差异,这些免疫细胞相关的分子标志物可能作为COVID-19免疫治疗的潜在靶标。
