Plastid genome evolution of a monophyletic group in the subtribe Lauriineae (Laureae,Lauraceae)
2022-09-01ChoLiuHunHunChenLiZhouTngPhyoKyKhineLiHongHnYuSongYunHongTn
Cho Liu ,Hun-Hun Chen ,Li-Zhou Tng ,Phyo Ky Khine ,Li-Hong Hn ,**,Yu Song ,Yun-Hong Tn,d,***
a College of Biological Resource and Food Engineering,Yunnan Engineering Research Center of Fruit Wine,Qujing Normal University,Qujing,Yunnan,655011,China
b Key Laboratory of Ecology of Rare and Endangered Species and Environmental Protection (Ministry of Education),Guangxi Key Laboratory of Landscape Resources Conservation and Sustainable Utilization in Lijiang River Basin,Guangxi Normal University,Guilin,Guangxi,541004,China
c Center for Integrative Conservation,Xishuangbanna Tropical Botanical Garden,Chinese Academy of Sciences,Mengla,Yunnan,666303,China
d Southeast Asia Biodiversity Research Institute,Chinese Academy of Sciences,Yezin,Nay Pyi Taw,05282,Myanmar
Keywords:Litsea Phylogenetic analysis Divergent hotspots Gene evolution
ABSTRACT Litsea,a non-monophyletic group of the tribe Laureae (Lauraceae),plays important roles in the tropical and subtropical forests of Asia,Australia,Central and North America,and the islands of the Pacific.However,intergeneric relationships between Litsea and Laurus,Lindera,Parasassafras and Sinosassafras of the tribe Laureae remain unresolved.In this study,we present phylogenetic analyses of seven newly sequenced Litsea plastomes,together with 47 Laureae plastomes obtained from public databases,representing six genera of the Laureae.Our results highlight two highly supported monophyletic groups of Litsea taxa.One is composed of 16 Litsea taxa and two Lindera taxa.The 18 plastomes of these taxa were further compared for their gene structure,codon usage,contraction and expansion of inverted repeats,sequence repeats,divergence hotspots,and gene evolution.The complete plastome size of newly sequenced taxa varied between 152,377 bp(Litsea auriculata)and 154,117 bp(Litsea pierrei).Seven of the 16 Litsea plastomes have a pair of insertions in the IRa (trnL-trnH) and IRb (ycf2) regions.The 18 plastomes of Litsea and Lindera taxa exhibit similar gene features,codon usage,oligonucleotide repeats,and inverted repeat dynamics.The codons with the highest frequency among these taxa favored A/T endings and each of these plastomes had nine divergence hotspots,which are located in the same regions.We also identified six protein coding genes(accD,ndhJ,rbcL,rpoC2,ycf1 and ycf2)under positive selection in Litsea;these genes may play important roles in adaptation of Litsea species to various environments.
1.Introduction
The genus Litsea Lamarck in the family Lauraceae,which is regarded as the‘core Laureae’or Litsea complex,is one of the most diverse genera of evergreen and rare deciduous trees or shrubs(Kamle et al.,2019).Litsea comprises approximately 400 known species and is distributed in the tropical and subtropical forests of Asia,Australia,Central and North America,and the islands of the Pacific (Fijridiyanto and Murakami,2009a).Litsea species are economically important as a source of medicine,spices,and perfumes (Wang et al.,2016),and serve as food for Muga silk worms(Antheraea assama) (Yadav and Goswami,1990).In addition,Litsea grow in mixed wood forest or forest edge,some of which used as garden trees,which play a critical role in ecoregulation.The genus Litsea was divided by Bentham (1880) into four sections: sect.Conodaphne(Bl.)Benth.et Hook.f.,sect.Cylicodaphne(Nees)Hook.f.,sect.Litsea,and sect.Tomingodaphne(Bl.)Hook.f.However,more recent studies have found that several Litsea species are nested within different sections.For example,some species of section Conodaphne are more closely related to Lindera than to other Litsea sections(Fijridiyanto and Murakami,2009b).In addition,Litsea and Lindera have been shown to be anatomically and morphologically polyphyletic,and closely related to Laurus,Dodecadenia Nees,Iteadaphne Blume,Parasassafras D.G.Long and Sinosassafras H.W.Li(Li and Christophel,2000;Li et al.,2008;Chanderbali et al.,2001;Song et al.,2020;Zhang et al.,2021).Delimiting the genera of the Litsea complex and understanding their phylogenetic relationships remains a major challenge.
Plastid and nuclear DNA barcoding markers have been used to provide effective molecular information for identification and comparison of closely related species or genera (Li et al.,2004;Fijridiyanto and Murakami,2009b;Song et al.,2016,2017,2020).Using the chloroplast gene matK and nuclear ribosomal DNA ITS sequences,Li et al.(2004)conducted a phylogenetic analysis of the‘core’ Laureae based on Chinese materials,and found that Litsea clustered with Laurus,Parasassafras,and Lindera in a polytomy.The phylogenetic analyses of rpb2 showed Litsea and Lindera were polyphyletic (Fijridiyanto and Murakami,2009b).Although different phylogenetic relationships among core Laureae have been investigated using chloroplast markers combined with nuclear ribosomal genes in previous studies(Li et al.,2004,2008;Fijridiyanto and Murakami,2009b),both the generic delimitation and species relationships within the genus Litsea still remain ambiguous.At present,high-throughput sequencing approaches might provide a better understanding of the relationships among species and genera in Lauraceae.
In recent years,complete plastid genomes have been widely used to study plant taxonomy and evolution in the family Lauraceae(Song et al.,2016,2017,2020;Tian et al.,2019;Xiao et al.,2020).Plastomes are ideal molecular markers for species identification and phylogenetic analysis because they are haploid,maternally inherited,and have a highly conserved structure,low mutation rates and a slow evolutionary rate(Du et al.,2020;Henriquez et al.,2020;Zheng et al.,2020).Comparative analysis of plastome structure and genetic diversity can help delimit genera and clarify phylogenetic relationships within the Lauraceae family.Currently,the plastome sequences of over ten Litsea species are available(Hinsinger and Strijk,2017;Hinsinger and Strijk,2017;Jo et al.,2019;Liu et al.,2020;Xiao et al.,2020).
Previous studies have used Lauraceae plastome sequences for phylogenetic analysis of Lauraceae (see Song et al.,2020,which reconstructed six monophyletic groups as the supergeneric classification of the Lauraceae).The tribe Laureae has been divided into at least five subgroups and Litsea taxa grouped into two subclades(Song et al.,2020).The first monophyletic group is composed of trinerved Lindera species and Iteadaphne caudata(Tian et al.,2019).In this study,we sequenced seven Litsea plastomes and compared the plastome structures of Litsea and Lindera taxa to identify the second monophyletic group in the Subtribe Lauriinae.This monophyletic group is composed of Litsea species and Lindera obtusiloba.We also identified divergence hotspot regions within the newly sequenced plastomes and detected protein coding sequences under positive selection.
2.Materials and methods
2.1.Sampling,DNA extraction and sequencing
Fresh young leaves of seven taxa (Litsea auriculata S.S.Chien &W.C.Cheng,Litsea dilleniifolia P.Y.Pai&P.H.Huang,Litsea lancilimba Merr.,Litsea liyuyingi H.Liu,Litsea pierrei Lecomte,L.pierrei var.szemois Liou,Litsea cubeba var.formosana (Nakai) Y.C.Yang &P.H.Huang)were obtained and dried in silica gel.The harvested tissues were deposited at the Biodiversity Research Group of Xishuangbanna Tropical Botanical Garden,Chinese Academy of Sciences (Table 1).Total genomic DNA was extracted from the leaves following the cetyltrimethylammonium bromide (CTAB)protocol(Doyle and Dickson,1987).Long-range PCR was performed following Zhang et al.(2016),with their 15 universal primer pairs for next-generation sequencing.A total of 6 μg of DNA was further fragmented into small pieces by Covaris S220,and paired-end libraries were constructed following the manufacturer's instructions(Illumina,San Diego,CA,USA).Sequencing was performed on an Illumina Genome Analyser Hiseq 2000 at Beijing Genomics Institute(Shenzhen,China).
2.2.Plastome assembly and annotation
Raw reads were filtered to remove low-quality reads,and de novo assembly of circular plastomes was carried out using GetOrganelle software (Jin et al.,2020).Plastomes were adjusted and annotated using Geneious v.8.1.3 (Kearse et al.,2012) and GeSeq(https://chlorobox.mpimp-golm.mpg.de/geseq.html) against the referenced plastid genome sequence of Litsea cubeba(Accession No.MT431385).Manual adjustment was carried out with published plastomes as a reference to confirm the boundaries between large single copy(LSC),small single copy(SSC),and inverted repeat (IR)regions.The precise locations of start and stop codons and the exons and introns of genes were carefully assigned manually.The output GenBank files were inspected and edited manually,and circular plastome maps were drawn using OGDRAW v.1.3.1(Greiner et al.,2019).
2.3.Phylogenetic analysis
To determine phylogenetic relationships of the “core Laureae”species,we compared the complete plastomes of 54 taxa of Lauraceae species available at NCBI and LCGDB(http://lcgdb.wordpress.com)(Song et al.,2020;Xiao et al.,2020)(Table S1),including two taxa each from Iteadaphne and Parasassafras,two from Laurus,21 from Litsea,27 from Lindera.We used two taxa from Cinnamomum Schaeff.as outgroups.

Table 1Summary of plastomes features of seven Litsea taxa.
Coding sequences (CDS) were also extracted and used for phylogenetic reconstruction.Multiple sequence alignments were initially done using MAFFT v.7.450(Katoh et al.,2019)and manually edited where necessary with BioEdit v.7.0.9.Bayesian inference(BI)was performed using BEAST v.2.6.3(Bouckaert et al.,2014)and the best substitution model (‘TVM+F’) was tested by AIC in IQ-TREE v.2.1.1 (Minh et al.,2020).Maximum likelihood (ML) analysis was performed using IQ-TREE(Minh et al.,2020)with the GTR+F+R2 model of nucleotide substitution and 1000 bootstrap replicates.
2.4.Sequence analysis
To assess evolutionary changes in the newly sequenced Litsea plastomes,we characterized plastome structure,including contractions and expansions of IR regions,and analyzed codon usage,repeats,and SSRs.The relative synonymous codon usage (RSCU)was obtained using CodonW v.1.4.2 (http://codonw.sourceforge.net/) with protein-coding genes >200 bp in size.GC content of the complete plastomes and the coding sequences were analysed using Mega X(Kumar et al.,2018).Long repetitive sequences were identified using REPuter web-service program (https://bibiserv.Cebitec.uni-bielefeld.de/reputer) (Kurtz et al.,2001).Forward(F),reverse (R),complementary (C),and palindromic (P) repeats were analysed with a minimum repeat size of 30 bp,a sequence identity of 90% and a Hamming distance of 3.Simple sequence repeats(SSRs)were detected using the MISA-web service(https://webblast.ipk-gatersleben.de/misa/) (Beier et al.,2017).SSRanalysis was employed with a minimum threshold of ten nucleotides for mononucleotides,five for dinucleotides,four for trinucleotides,and three for tetranucleotides,pentanucleotides,and hexanucleotides.The nucleotide diversity (Pi) value was calculated through alignment of the complete plastome sequences of different species using DnaSP v.6.0 (Rozas et al.,2017),with the window length set to 600 bp and a 200-bp step size.Values were plotted in the R program.Gene organization maps were drawn in TBtools v.1.064 (Chen et al.,2020).Expansion and contraction of IRs was analysed using the IRscope online program (https://irscope.shinyapps.io/irapp/).To evaluate variation in evolutionary rates of plastomes,we examined 82 protein-coding regions in 18 taxa belonging to one monophyletic group of sect.Litsea(16 Litsea species and 2 Lindera species).The site model was performed to test for potential positive selection using the CODEML algorithm in PAMLX v.1.3.1 under a phylogenetic framework (Xu and Yang,2013).The ratios of non-synonymous(dN) to synonymous (dS) substitutions (dN/dS) in proteincoding genes were estimated.Two likelihood ratio tests (LRT) of M1 (nearly neutral) vs.M2 (positive selection) and M7 (beta) vs.M8 (beta and ω) were used to check for sites with evidence of positive selection.The posterior probabilities for evolutionary potential under selection were calculated using the Bayes empirical Bayes (BEB) method.The species Litsea cubeba(MT431385) was used as a reference.
3.Results
3.1.Characteristics of the seven newly obtained Litsea plastomes
In this study,we sequenced seven Litsea plastomes.Plastome sizes ranged from 152,377 bp(L.auriculata)to 154,117 bp(L.pierrei)(Fig.1).The typical quadripartite structure was found for all seven taxa: a pair of IRs (IRa and IRb) of 20,015-21,289 bp separated by one LSC region of 92,643-94,311 bp and one SSC region of 18,333-18,814 bp.The plastomes of two species(L.dilleniifolia and L.pierrei var.szemois)lost a 660-bp fragment between two poly-A/T sequences.GC content was similar in all seven plastomes(39.15%-39.24%).GC content (43.96%-44.70%)in the IR regions was higher than that in the LSC (37.93%-38.05%) and SSC regions (33.87%-34.12%).GC content in protein-coding genes was 39.36%.A total of 113 single copy genes were detected in each plastome,including four rRNA genes,30 tRNA genes,and 79 protein-coding genes.In addition,13 duplicated genes were located in the IR regions(Tables 1 and S2).Among the protein coding genes,nine genes(atpF,ndhA,ndhB,petB,petD,rpl2,rpl16,rpoC1 and rps16) displayed one intron each;three genes(clpP,rps12 and ycf3)contained two introns each.
3.2.Phylogenomic analysis
To understand the evolutionary relationships between Litsea taxa,we reconstructed a Laureae phylogenetic tree using the plastomes of 54 Lauraceae taxa,including our seven newly sequenced Litsea plastomes and plastomes of 14 additional Litsea taxa.The phylogenetic trees inferred from ML and BI analyses were topologically congruent,and the phylogenetic topology of trees based on complete plastomes and coding sequences was the same,with similar values of branch support(Figs.2 and S1).Thus,here we only show the Bayesian tree based on complete plastomes.The 54 Lauraceae taxa were divided into at least four clades with strong support,excluding the two Cinnamomum species,which served as outgroups.Clade I included two Laurus,seven Lindera,and five Litsea taxa;Clade II included four Lindera species;Clade III included two Lindera,and 16 Litsea taxa;Clade IV included one Iteadaphne,one Parasassafras,and 14 Lindera taxa.The 21 Litsea taxa were clustered into two clades (Clade I and Clade III),and the seven newly obtained Litsea taxa were located in the same sub-clade with an additional nine Litsea and two Lindera obtusiloba taxa.Clade III was split into four major subclades: clade IIIa (Litsea cubeba var.formosana,L.mollis,L.cubeba,L.panamonja,Lindera obtusiloba var.heterophylla,L.obtusiloba),clade IIIb(L.auriculata,L.coreana),clade IIIc(L.elongata,L.japonica,L.garrettii,L.monopetala),and clade IIId(L.dilleniifolia,L.szemaois,L.pierrei,L.pierrei var.szemois,L.liyuyingi,L.lancilimba).
3.3.Codon usage and amino acid frequency
In clade III,each of the 18 plastomes possessed 82 proteincoding genes,which were encoded by 64 different codons,including three stop codons (TAA,TAG and TGA,Fig.3).RSCU analyses revealed that codons that ended with A/T at the third base had RSCU>1 and encoded the highest number of amino acids.The codons that ended with C/G at the third base had RSCU <1 and encoded the lowest number of amino acids(Fig.3).Most of the high frequency codons and 29 out of 31 optimal codons ended with A/T.Among the 64 codons,AGC-encoded serine and CGC-encoded arginine had the lowest RSCU values (0.35),and AGA-encoded arginine had the highest RSCU values (1.82).The ATG codon,which encodes formyl-methionine as a start codon,was the most common start codon in this study.However,alternate codons were also found as start codons,such as GTG(in cemA and rps19),TTG(in ndhD and rpl2),CTG (in psbL) and ACT (in rps12),which has also been reported in other plant species(Henriquez et al.,2020;Zheng et al.,2020).Moreover,the analysis of amino acid frequencies revealed that leucine (Leu,10%) and cysteine (Cys,1%) were the most and the least frequent amino acids,respectively (Fig.S2).In general,codon usage and amino acid frequency were similar across the 18 taxa.

Fig.1.Gene map of the Litsea plastomes. Litsea garrettii is shown.
3.4.Contraction and expansion of inverted repeats
Contractions and expansions of inverted repeats at the junctions of LSC/IRb,IRb/SSC,SSC/IRa,and IRa/LSC revealed that the plastomes of the 18 taxa in clade III are highly similar.L.pierrei var.szemois has the longest IR region(21,289 bp),whereas L.auriculata has the shortest(20,015 bp),with a difference of 1274 bp.Thus,the lengths of the IR regions of the 18 taxa are very different.The ycf2 gene spans the LSC/IRb boundary,and partially extends into the IRb region that ranges from 3159 bp to 3863 bp in length.The ndhF gene spans the IRb/SSC boundary,and the portion located in the IRb region ranges from five to 12 bp in length.The ycf1 gene spans the junction of the SSC and IRa regions,and the length of the ycf1 gene extends into the IRa region,ranging from 5456 bp to 5588 bp.Finally,the trnH gene is completely located in the LSC region at the junction of the IRa/LSC,and its distance from the boundary line is between 21 bp to 27 bp.Overall,the 18 taxa show similar characteristics at the IR/SC boundary regions.The LSC and SSC regions are more divergent between species than the two IR regions,and the intergenic regions exhibit higher divergence than the coding regions.In the alignment analysis,seven regions (trnG-trnR,psbMtrnD,ycf3-trnS,ndhF-rp132-trnL and ycf1) with low identity were detected (Fig.S3).The plastomes of seven Litsea species(L.dilleniifolia,L.elongata,L.garrettii,L.lancilimba,L.pierrei,L.pierrei var.szemois,L.cubeba var.formosana) have a 678-bp insertion in the IRa (intergenic region) and IRb (ycf2) regions,enlarging their genome size (Table 1;Fig.S4).The gene order between the two IR regions is highly conserved in the plastomes of the 18 tax of clade III.
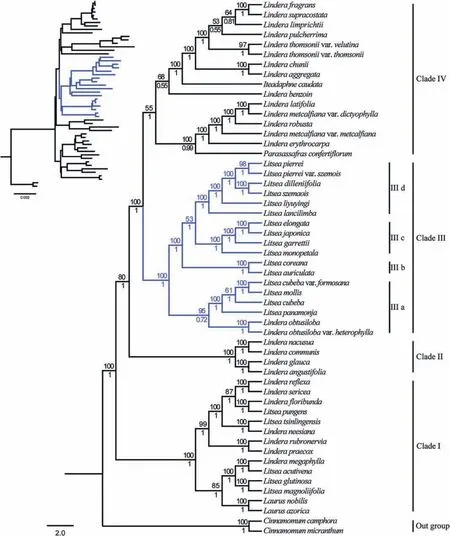
Fig.2.Bayesian tree of 54 Laureae based on plastome sequence data.Numbers above nodes are bootstrap values.Blue branches represent the 18 taxa analysed in this study.
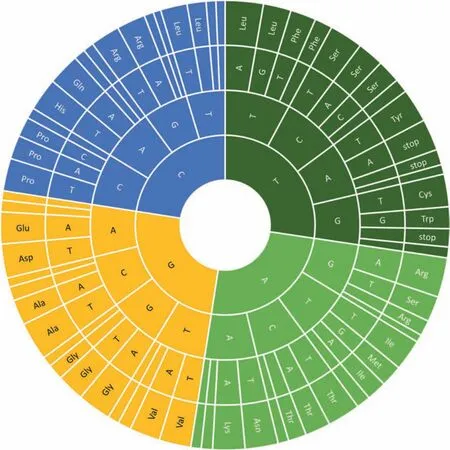
Fig.3.Codon-anticodon recognition patterns and codon usage in plastomes of the 18 taxa.
3.5.Repeat analysis
Four types of oligonucleotide repeats — palindromic,reverse,forward,and complement repeats — were detected in the plastomes of the 18 taxa in clade III(Fig.4).A total of 555 long repeats,consisting of 226 forward repeats and 217 palindromic repeats,were detected (Fig.4A).The forward,palindromic and reverse repeats were abundant in L.japonica,L.dilleniifolia,and L.cubeba var.formosana,respectively.The size of repeats varied among species,but most of the repeat sizes were in the range of 30-34 bp(Fig.4B).Among the 18 taxa,L.pierrei var.szemois and L.garrettii/L.monopetala had the largest (39) and smallest (24) number of repeats.The number of forward repeats varied between nine(L.garrettii) and 16 (L.japonica),and the number of palindromic repeats varied from 10(L.cubeba,L.cubeba var.formosana,L.mollis and L.panamonja) to 16 (L.dilleniifolia).The lengths of forward repeats in the 18 species ranged from 30 to 57 bp,and the lengths of palindromic repeats varied from 30 to 60 bp.The number,length and distribution of these sequences varied among the 18 taxa(Fig.4A).
3.6.SSR analysis
Due to high levels of polymorphism,SSRs are often used as genetic markers to identify closely related species.We compared the distribution and number of SSRs in the plastomes of the 18 taxa in clade III.SSRs ranging from mononucleotide to hexanucleotide repeats were found,but not in all species (Fig.5A).We identified 1537 SSRs from the 18 plastomes,including 1092 mononucleotide(71.05%),177 dinucleotide (11.52%),67 trinucleotide (4.36%),160 tetranucleotide (10.41%),21 pentanucleotide (1.37%),and 20 hexanucleotide(1.30%).The first four types of SSR repeats were present in each of the 18 taxa,while the pentanucleotide repeats were absent in L.pierrei var.szemois,and hexanucleotide repeats were absent in L.auriculata and L.liyuyingi.The maximum and minimum length of the SSRs was 27 bp(A/T in L.coreana),and eight bp(in all 18 taxa),respectively.On average,mononucleotide repeats were the most abundant,accounting for 71.05%of the SSRs,followed by dinucleotide repeats (11.52%) and tetranucleotide repeats (10.41%)(Fig.5B).L.monopetala and L.mollis had the most (95) and the fewest SSRs(77),respectively.There were no significant differences in the number of SSRs among the 18 taxa.
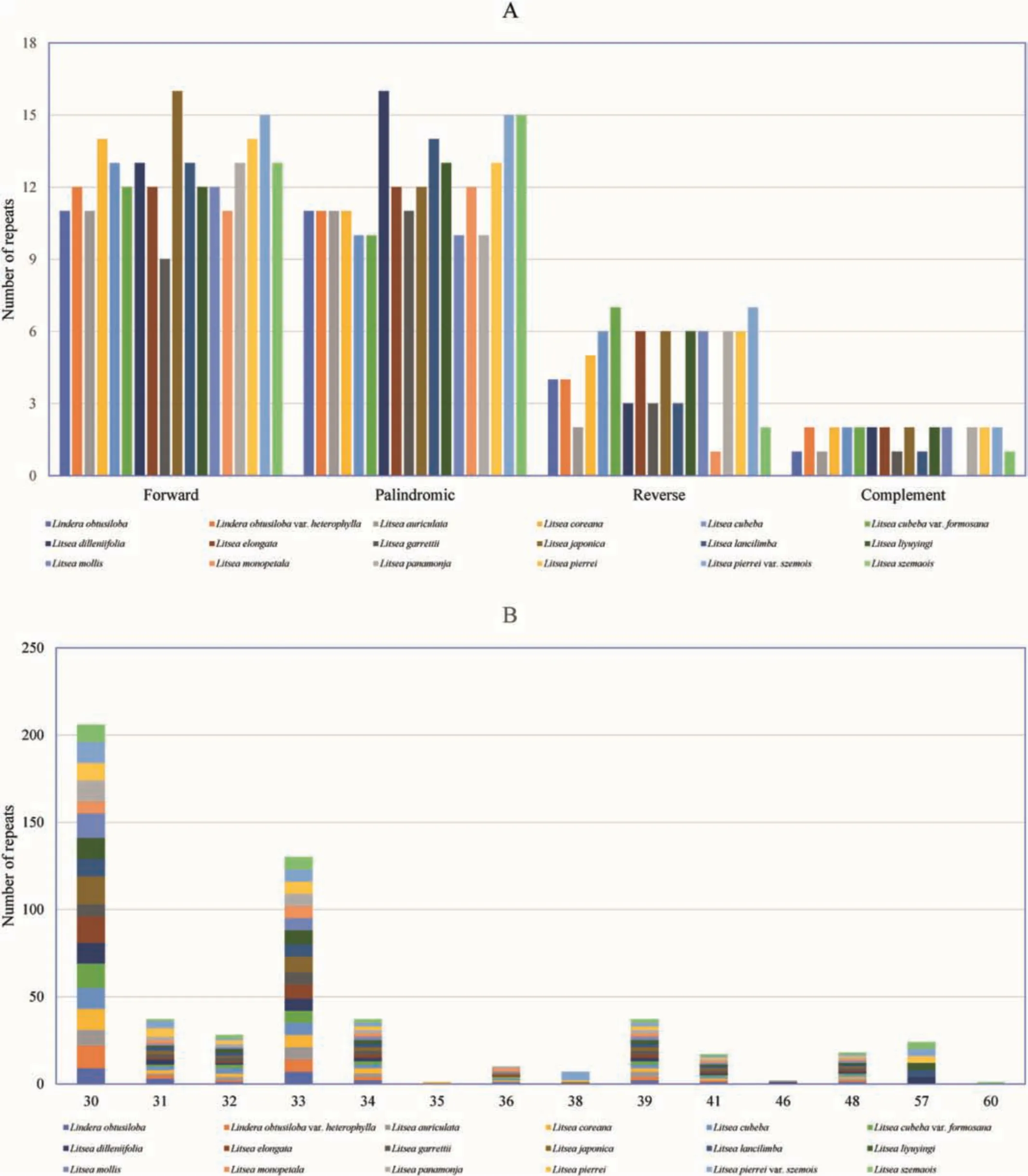
Fig.4.Number of long repetitive repeats in plastome sequences of the 18 taxa.A.Frequency of repeat type.B.Frequency of repeats >30 bp.

Fig.5.Number of SSRs in plastome sequences of the 18 taxa.A.Frequency of repeats >30 bp.B.Frequency of repeat type.
3.7.Divergence hotspots

Fig.6.Nucleotide variability (Pi) values in aligned plastomes from 18 taxa.
Nucleotide diversity (Pi) values were analysed by DnaSP to measure divergence levels within different plastome regions of the 18 taxa in clade III.The alignment size was 156,419 bp,of which 150,516(about 96.2%)sites were constant,and 2685(about 1.7%) nucleotide sites were polymorphic.Pi values ranged from 0 to 0.01854,and the average value was 0.00367(Fig.6).In the LSC region,Pi values ranged from 0.00019 to 0.01241,with an average of 0.00416,and in the SSC region,they ranged from 0.00277 to 0.01854,with an average value of 0.00749;the IR region,in which Pi values ranged from 0 to only 0.00534,had the lowest average value (0.00075) (Fig.6).Within the nine divergence hotspots,three regions(trnG-trnR,psbM-trnD and ycf3-trnS)were located in the LSC region and six regions(ndhF,ndhF-rpl32,rpl32-trnL,ndhGndhI,ycf1a,and ycf1b)were in the SSC region.No variation in the IR regions implies that these regions are more conserved than the SSC and LSC regions.The relatively higher divergence of ndhF,rpl32 and ycf1 genes than other coding regions has been previously observed in other plastomes (Zheng et al.,2020).
3.8.Selective pressure analysis of protein-coding genes
To test whether the plastid genes of the 18 taxa in clade III had undergone selection,we estimated the rate of dS,dN,and the ratio of dN/dS based on function classification(Fig.7).The dN/dS ratios were used to estimate the selective pressure in the context of a codon substitution model,with dN/dS <1,dN/dS=1,and dN/dS >1 denoting purifying,neutral,and positive selections,respectively.The dN/dS values were less than 1 for most genes(74 of 79;93.7%),indicating that purifying selection was acting on these genes.Five genes (rps16,petA,ndhC,rps3,and rpoA) in the analysed plastomes showed average dN/dS ratios higher than 1,indicating positive selection had been exerted on these genes.A total of 20 protein coding genes obtained feedback results based on BEB analysis(Table S3).Of these,nine genes contained at least one positively selected site with significant posterior probabilities,and rbcL,rpoA,rpoB,rpoC2,ycf1,and ycf2 possessed three or more significantly and positively selected sites.Six protein-coding genes (accD,ndhJ,rbcL,rpoC2,ycf1,and ycf2) rejected the null model (P <0.01),corroborating the hypothesis that some amino acid sites in these proteins had been under positive selection.Positive selection of these genes indicates that they are undergoing essential adaptations to their environment despite the weak selection pressures experienced by Litsea.
4.Discussion
4.1.Phylogenomic and comparative analysis
The systematics of the“core Lauraceae”group remain in dispute(Tian et al.,2019;Xiao et al.,2020).Previous studies have divided the “core Lauraceae” into two or five major clades based on complete plastome sequences (Zhao et al.,2018;Tian et al.,2019).Furthermore,representatives of the sections of Litsea genera have been described as polyphyletic (Li et al.,2008).Here,we used complete plastome sequences of 54 Laureae species to determine the phylogenetic positions of Laureae species.In contrast to previous studies,we found that the “core Lauraceae” can be divided into four clades with strong support.Sixteen of 21 Litsea taxa and two Lindera taxa clustered in clade III,forming a monophyletic lineage.The genus Litsea can be divided into four sections,as proposed by Bentham (1880),with L.coreana and L.monopetala belonging to section Conodaphne;Litsea glutinosa belonging to section Litsea;L.auriculata,L.cubeba,L.cubeba var.formosana,L.mollis,L.pungens,and L.tsinlingensis belonging to section Tomingodaphne;and the other eleven Litsea species (except for L.japonica)belong in to section Cylicodaphne.Clade I contains two Tomingodaphne species (L.pungens and L.tsinlingensis),two Cylicodaphne species(Litsea magnoliifolia and Litsea acutivena),and one Litsea species (L.glutinosa),while Clade III includes four Tomingodaphne taxa (L.auriculata,L.cubeba,L.cubeba var.formosana,and L.mollis),two Conodaphne species (L.coreana and L.monopetala),and nine Cylicodaphne species.
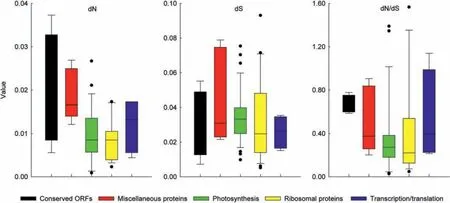
Fig.7.Positive selection strength of protein-coding genes of the plastomes from 18 taxa.
The plastid gene content of land plants does not appear to have changed dramatically,and few gene losses might have taken place during the evolution of land plants (Wicke et al.,2011;Song et al.,2017).Available plastomes of the Lauraceae have similar structures and vary in size from 150 kb to 159 kb,except for genus Cassytha(115 kb),which has lost an entire IR region (Song et al.,2017).Throughout the evolution of terrestrial plants,the IR regions have shown a tendency to expand(Zheng et al.,2020).In this study,we report complete plastome sequences of seven Litsea taxa in the tribe Laureae (Lauraceae).The complete plastome size of newly sequenced taxa varies from 152,377 bp(L.auriculata)to 154,117 bp(L.pierrei).IR region lengths of the 18 taxa of clade III also vary(20,015-21,289 bp) and are shorter than those of Lagerstroemia(Zheng et al.,2020) and Monsteroideae (Henriquez et al.,2020).This variation in IR region length differs from that of other species of Lauraceae (Tian et al.,2019;Xiao et al.,2020).In Litsea species,variation of plastome size can be largely explained by expansions and contractions of the IR regions.Unlike other Lauraceae plants,the plants of clade III have identical numbers of protein-coding genes (82),transfer RNA genes (tRNA) (36) and ribosome RNA(rRNA) genes (8) (Song et al.,2016,2017).In contrast to previous studies(Xiao et al.,2020),we found that the plastomes of Lauraceae plants have more rpl22 and psbZ genes than lhbA genes.
4.2.Sequence repeats and divergence hotspot analysis
Oligonucleotide repeats play important roles in the generation of substitutions,indels,and inversions (Iram et al.,2019).In this study,analyses of oligonucleotide repeats and SSRs showed similarities among plastomes of the 18 taxa.Most of the repeats ranged between 30 and 34 bp.The most frequent mononucleotide repeats were A/T repeats (97.7%);the remaining 2.3% of mononucleotide repeats were G/C repeats.High proportions of poly A/T have also been reported in previous studies (Zheng et al.,2020;Mehmood et al.,2020;Munyao et al.,2020).Three species (L.dilleniifolia,L.szemaois,L.pierrei var.szemois) have a 660-bp indel flanked by poly-A/T repeats.
Divergence hotspots can provide references for the development of accurate,robust,and cost-effective molecular markers for phylogenetic analysis,population genetics and species barcoding(Ahmed et al.,2013).In the 18 plastomes of clade III the regions with the highest nucleotide variability include trnG-trnR,psbMtrnD,ycf3-trnS,ndhF,ndhF-rpl32,rpl32-trnL,ndhG-ndhI,and ycf1.The highly variable hotspots screened in this study exhibit divergence similar to that of psbM-trnD,ndhF,and ycf1 in trinerved Lindera species (Tian et al.,2019),and of ndhF-rpl32,and ycf1 in nine Lindera species (Zhao et al.,2018).Unlike Lindera,the Litsea species showed hypervariable regions at the locus of trnG-trnR,ycf3-trnS,rpl32-trnL,and ndhG-ndhI.The mini-barcode ndhF-rpl32 also performed well in distinguishing Pterocarpus species (Jiao et al.,2018).The ndhF,ycf1,and rpl32 are highly variable in many plants and even used for barcoding (Amar,2020;Zheng et al.,2020).The highly variable hotspots and polymorphic SSRs in the study may provide references for marker-assisted breeding and identification of Litsea cultivars.
4.3.Codon usage and selective pressure analysis
Base composition has a substantial effect on the basic parameters of genome stability and evolution (Kiktev et al.,2018).In the genus Oryza,GC content correlates positively with recombination rates,and GC content at the 3′is positively correlated with expression levels,suggesting that selection favors codons ending with G or C (Muyle et al.,2011).Monocotyledons favor G or C as terminated codons,while dicotyledons favor A or T (Chen et al.,2017).In the current study,Litsea GC content is similar to that of Lindera species(Xiao et al.,2020),and the codons that end with A or T at the 3′show high encoding efficacy of the amino acid.The codon usage patterns of Litsea species are similar to those reported in Lagerstroemia and Monsteroideae,possibly due to the high proportion of A/T in plastomes (Zheng et al.,2020;Henriquez et al.,2020),which illustrates nucleotide composition plays a vital role in the pattern of genes codon usage.
Because recombination events are rare in the plastid genome,it has a low DNA replacement rate and genes are generally conserved(Omelchenko et al.,2020).Our analysis of Litsea plastomes shows that 74 of 79 genes have an average of dN/dS <1,which indicates that,as expected,the evolutionary rate is low in these species.Most of these genes,which are associated with photosynthesis,are crucial and conserved in plants (Liang et al.,2020);thus,no nonsynonymous mutations were detected.Six genes (accD,ndhJ,rbcL,rpoC2,ycf1,and ycf2) often associated with photosynthesis and gene transcription possess at least one site under positive selection in Litsea(Gao et al.,2019;Wang et al.,2021;Yang et al.,2021).The positive selection of rbcL gene,which encodes for the large subunit of RuBisCO,is fairly common in many land plants(Wen et al.,2021).In Ilex,high rates of positive selection on the rbcL gene are associated with hybridization and introgression (Yao et al.,2019).The acetyl-CoA carboxylase subunit D gene accD,NADH oxidoreductase gene ndhJ and the transcription gene rpoC2 have been shown to be under positive selection pressure in several plant species(Gao et al.,2019;Yang et al.,2021).The functions of ycf1 and ycf2 are largely unknown,but they are indispensable for plant cell survival(Drescher et al.,2000).These positively selected genes and nucleotide characteristics found in Litsea species may play an important role in helping these plants adapt to various environments.
5.Conclusions
In this study,the plastomes of seven Litsea taxa were sequenced for phylogenetic analyses.Our analysis showed that the genus Litsea is polyphyletic.Sixteen Litsea taxa clustered with two Lindera taxa in a monophyletic group.Our findings reveal the phylogenetic relationships of the Laureae species.Comparative analysis of the plastomes of 18 taxa of this monophyletic group revealed a conserved collinear structure and genetic content of the sequences,and provide reliable genetic information for the study of subtribe Lauriineae.We also identified SSR markers and highly variable regions that may serve as significant genetic markers to delimit species/cultivars and provide a reference for marker-assisted breeding.The positive selection of several genes (accD,ndhJ,rbcL,rpoC2,ycf1,and ycf2) may reflect the rapid evolution in some lineages of genus Litsea.
Author contributions
YS and CL conceived and designed the work,YHT,LZT and HHC prepared the datasets and discussed the results,CL wrote the manuscript,PKH,LHH and YS revised the manuscript.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant No.32060710,31970223,31860005,31860620) and Applied Basic Research Projects of Yunnan (Grant No.2019FB057).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.pld.2021.11.009.
杂志排行

植物多样性的其它文章
- V.PhyloMaker2:An updated and enlarged R package that can generate very large phylogenies for vascular plants
- Recent advances on phylogenomics of gymnosperms and a new classification
- Phylogenetic endemism of the orchids of Megamexico reveals complementary areas for conservation
- Ontogenetic trait variation and metacommunity effects influence species relative abundances during tree community assembly
- Reproduction and genetic diversity of Juniperus squamata along an elevational gradient in the Hengduan Mountains
- Parahellenia,a new genus segregated from Hellenia (Costaceae) based on phylogenetic and morphological evidence