Molecular pathology of thymoma and thymic carcinoma
2022-07-20DavidSusterMalayKumarBasuCraigMackinnonDepartmentofPathologyRutgersUniversityNewJerseyMedicalSchoolNewarkNJ0703USA
David I. Suster, Malay Kumar Basu, A. Craig MackinnonDepartment of Pathology, Rutgers University New Jersey Medical School, Newark, NJ 0703, USA.
2Department of Pathology, University of Alabama at Birmingham, Birmingham, AL 35233, USA.
Abstract Thymic epithelial tumors (TETs) comprise a heterogeneous group of epithelial-derived thymic neoplasms with diverse clinical behavior and underlying molecular genetic features. Owing to their rare nature, the molecular classification of TETs has only recently begun to be fully explored. The advent of advanced molecular studies,particularly the ability to sequence the DNA and RNA of tumors in a massively parallel fashion, has led to an increased understanding of the molecular underpinnings of thymic neoplasia. Thymomas, characterized by a heterogeneous group of molecular alterations, tend to have low mutational burdens and various copy number abnormalities including a characteristic loss of chromosomal material in the region of 6q25.2-p25.3, a recurrent,specific point mutation GTF2I p.L424H, and specific expression of certain microRNAs. Thymic carcinomas, in contrast, are generally characterized by increased tumor mutational burdens, multiple copy number alterations,and varied, non-recurrent, somatic mutations. Advances in molecular knowledge of TETs allow for more precise molecular classification of these tumors, and the presence of specific alterations aids in the diagnosis of borderline lesions. In the future, additional molecular studies will better delineate the molecular landscape of these tumors and may one day allow for more targeted treatment algorithms. This review aims to cover the current understanding of the molecular alterations thus far identified in thymomas and thymic carcinomas.
Keywords: Thymoma, thymic carcinoma, GTF2I, molecular, genetics
INTRODUCTION
Thymic epithelial tumors (TETs) comprise a heterogeneous group of epithelial-derived thymic neoplasms with diverse clinical behavior and underlying molecular genetic features[1]. Thymomas and thymic carcinomas are the most common group of epithelial tumors arising within the mediastinum and represent a rare occurrence relative to other human cancers. They are estimated to have an incidence of 1.3 per 1,000,000 person-years, with only approximately 400 new cases diagnosed per year in the United States[2].Thymomas make up the majority of lesions, with thymic carcinomas occurring only rarely. Even less commonly, epithelial-derived tumors with neuroendocrine differentiation or salivary gland type features may occur within the mediastinal compartment[3].
Traditionally and in current clinical practice, TETs are classified primarily based on histologic features.Numerous classification systems have been proposed and used over the years, with the World Health Organization (WHO) classification system being the most widely accepted[1,4-8]. Despite the presence of numerous histology-based classification systems, problems still exist with the most current classification schema due to the varied biological behavior of TETs[4-9]. It is known that in some instances, the clinical and biological behavior may differ between tumors with similar histology[9]. In addition, problems with interobserver reproducibility lead to difficulties in classifying tumors and predicting their biological behavior.
Staging of TETs is somewhat more simplified; the most commonly used staging is the modified Masaoka staging system, although more recently, a tumor-nodes-metastases system has been adopted by the American Joint Committee on Cancer in the 8th edition of their cancer staging manual[10-12]. However, even within the confines of these consolidated staging systems, tumors within the same stage category may still sometimes display varied biological behavior[13].
Owing to their rare nature, the molecular classification of TETs has only recently begun to be fully explored.The advent of advanced molecular studies, particularly the ability to sequence the DNA and RNA of tumors in a massively parallel fashion, has led to an increased understanding of the molecular underpinnings of thymic neoplasia. The varied biologic behavior of thymic epithelial neoplasms, including histologic classification and staging discrepancies, is likely explained by their underlying molecular features. This review evaluates the currently known molecular information, primarily focusing on thymomas and thymic carcinomas, and describes some of the evolving molecular classifications of these tumors that may one day lead to an integrated morpho-molecular classification that will better prognosticate these tumors and allow for potential future targeted therapies.
MOLECULAR FEATURES OF THYMOMA AND THYMIC CARCINOMA
Molecular landscape of thymoma
Thymomas are rare tumors that occur predominantly in the 5th and 6th decades of life, with an estimated incidence of approximately 15-30 cases per 100,000 people per year[14,15]. Currently, thymomas are classified based primarily on their histomorphologic features. The most commonly used classification system is that of the WHO, which classifies thymomas into five distinct subtypes based on the type of cell (spindlevs.epithelioid) and the relative proportion of associated lymphocytes[1]. The five distinct subtypes include type A (predominantly spindled), type AB (spindled with increased lymphocytes), type B1 (sparse epithelioid cells with abundant lymphocytes), type B2 (increased epithelioid cells with relatively fewer lymphocytes),and type B3 thymoma (epithelioid cells with atypical features and few lymphocytes) [Figure 1][2]. It is worth noting that this letter-number classification was originally meant to simply serve as a method for translating the prior Bernatz and Muller-Hermelink classification and lends itself to problems with reproducibility and is generally insufficient at classifying many of the distinct subtypes of thymoma that exist. Type A and AB tumors tend to present in lower clinical stages as compared to B1/B2/B3 thymomas[16]. A unique clinical characteristic of these tumors is their association with paraneoplastic syndromes, the most common of which is myasthenia gravis. Myasthenia gravis may occur in up to 25%-45% of thymoma patients and more commonly occurs in patients with lymphoid-rich tumors such as AB and B types[17]. Various other paraneoplastic syndromes including various immunodeficiency and autoimmunity states have been described and may be related to the underlying molecular biology of these tumors[18-20].

Figure 1. WHO histologic classification of thymoma (A) Type A thymoma is characterized by a spindle cell proliferation with limited numbers of lymphocytes, (B) Type AB thymoma is characterized by a spindle cell population intimately admixed with increased numbers of lymphocytes, (C) Type B1 thymoma is characterized by sparse epithelioid cells in a dense immature lymphoid cell population, (D) Type B2 thymoma is characterized by an increased number of epithelioid cells with a relatively decreased proportion of lymphoid cells, (E) Atypical thymoma (WHO type B3 thymoma) is characterized by sheets of epidermoid appearing epithelioid cells with increased cytologic atypia and few lymphocytes, (F) Metaplastic thymoma is characterized by a pseudosarcomatous stromal component with an admixed epithelial component composed of neoplastic thymic epithelial cells. Note: Not shown here are numerous other subtypes of more esoteric thymomas that do not fit into any particular classification system and have limited documented molecular data.
Owing to their rarity, the molecular characteristics of thymoma have lagged behind other more common tumor types. Currently, prognostication is based primarily upon post-surgical staging of the tumors;however, the lack of a more refined classification and prognostication system leads to difficulty in the management and treatment of patients with thymoma. In the past few decades, numerous studies have begun unraveling the molecular landscape of thymomas. Inoueet al.[21]performed an extensive molecular study of multiple thymoma subtypes utilizing comparative genomic hybridization and analysis of microsatellite markers across numerous chromosomes. This study identified an increased number of genetic aberrations including frequent and multiple loss of heterozygosity across chromosome 6 (the 6q25.2-p25.3 region contains theFOXC1tumor suppressor gene) in thymoma types A, AB, B2, and B3.Type B1 thymoma was not included in the study. In addition, frequent allelic imbalances were identified on chromosomes 13q14.3 (RBgene), 16q22.1 (CDH1gene), 8p11.21, and 7p15.3, primarily within non-A subtypes of thymoma. These alterations were found to generally correlate with tumor stage, with alterations at 8p11.21 and 16q22.1 occurring more frequently in stage IV thymomas. Numerous other cytogenetic abnormalities have also been identified in the various subtypes of thymoma, with types AB and B2 having multiple reported cases that demonstrate complex karyotypes as well other scattered cytogenetic alterations including rare translocations, ring chromosomes, and various copy number alterations[22-32].
More recently, advanced molecular studies and multi-omic studies have identified additional molecular features of thymomas. This data can be used to further stratify these lesions based on their genetics, and some genetic changes are so highly recurrent that they are being used as diagnostic biomarkers. Both Enkneret al.[33]and Radovichet al.[34]identified the expression of multiple microRNAs (miRNA) including overexpression of miRNA clusters on chromosomes 19q13.42 and 14q32 that can be regarded as transcriptional hallmarks of WHO type A and AB thymomas. In addition, multiple somatic mutations have been identified scattered throughout the spectrum of thymomas. Perhaps the most important of these is a recurrent somatic mutation in the general transcription factor II-I (GTF2I) gene, which is mutated in approximately 80%-100% of type A thymomas, 70%-80% of type AB thymomas[35,36]and less commonly (0%-30%) in type B thymomas and thymic carcinoma. This mutation occurs on chromosome 7 and results in a T> A transversion resulting in a missense mutation that exchanges histidine for leucine at amino acid position 424 (position chr7:74732629 on build38). The mutation is annotated asGTF2Ip.L424H [Figure 2].Somatic mutations across theGTF2Igene are documented in human cancers including other types of epithelial tumors, central nervous system tumors, and hematologic malignancies; however, these are rare and the L424H variant thus far appears only to occur in thymic epithelial neoplasms. The increased incidence of this mutation in the more indolent thymoma subtypes (A1 and AB) indicates that this mutation harbors positive prognostic significance in these tumors. Furthermore, due to its specificity for thymic epithelial neoplasms,GTF2Ip.L424H is diagnostically useful to differentiate thymic neoplasms from other entities that enter the histologic differential. Somatic mutations occurring in other genes, such asHRAS,NRAS, andTP53, have also been described, but these tend to be rare and provide less prognostic or diagnostic utility[37]. Finally, it is worth noting that gene fusions may rarely occur in thymic epithelial neoplasms. RecurrentYAP1-MAML2gene fusion events were recently identified in metaplastic thymoma -a specific subtype of thymoma which does not fit into any of the conventional WHO categories[38]. These fusions were identified in treatment-naïve tumors indicating that they are the primary driver alterations in these tumors. A microdissection study using FISH demonstrated that the rearrangement can be identified within the epithelial and spindle cell components of these tumors, suggesting that the spindled component shares a common lineage with the epithelial component[39]. Although not yet targetable,YAP1-MAML2appears to be fairly specific for metaplastic thymoma[38,39]. RareKMT2A-MAML2gene fusion events have been described in a small percentage of type B2 and B3 thymomas and are thought to correlate with more aggressive behavior; of note, this rearrangement was originally identified in a tumor that had undergone induction therapy, suggesting it may represent a secondary therapy-related genetic event[40]. Future studies may yet uncover additional fusion genes that play a role in the tumorigenesis of these lesions.

Figure 2. Lollipop plot from St. Judes PECAN database demonstrating somatic point mutations across the GTF2I gene. GTF2I gene(transcript RM_032999) is shown with overlayed data from the COSMIC database. Each individual dot corresponds to a documented mutation. The majority of the mutations are of missense type (blue dots). GTF2I p.L424H is one of the most commonly occurring mutations. Source of data and image: https://pecan.stjude.cloud/ and https://cancer.sanger.ac.uk/cosmic[77-79].
Molecular classification of thymoma
In the past few years, two significant molecular studies have been published that describe potential molecular classification systems for thymomas. The first study by Leeet al.[31]stratified thymomas into four separate molecularly defined groups: (1) theGTF2Igroup (enriched forGTF2Imutations); (2) the T-cell signaling group characterized by increased expression of genes related to T-cell signaling; (3) the chromosomal stability group characterized by low levels of chromosomal abnormalities, and (4) the chromosomal instability group characterized by increased genetic complexity[31]. Interestingly, these molecular groups roughly correlated with traditional histologic classification and staging parameters: type A and AB thymomas tended to fall into theGTF2Igroup, type B1 and B2 thymomas tended to fall into the Tcell signaling group, and type B3 thymomas tended to fall into the chromosomal instability group.
A second large, integrated molecular analysis performed by Radovichet al.[37]led to a four-tiered molecular classification system in which thymomas were assigned to one of three molecular subtypes, and thymic carcinoma was assigned to the fourth group. The molecular thymoma subtypes include an A-like cluster and AB-like cluster that share many similarities including frequentGTF2Imutations and overexpression of the C19 miRNA cluster; these two groups are distinguished by differences in regulation ofp53, MYB, MYC,andFOXM1. The third molecular thymoma subtype is the B-like cluster characterized by the absence of bothGTF2Imutations and overexpression of the C19 miRNA cluster. The B-like subtype is also more closely associated with myasthenia gravis. This study also demonstrated that thymomas generally have low mutational burdens. We recently tested a small cohort of atypical type B3 thymomas using a 50 gene nextgeneration sequencing panel and similarly found a low number of non-synonymous mutations indicating that even some cases of the more aggressive subtype of thymomas harbor a low number of somatic mutations, although these tumors would benefit from testing with more extensive panels to more clearly delineate this [Figure 3]. A comparison of the aforementioned proposed molecular classification systems is presented in Table 1. More recent studies have identified additional gene expression profiles within different thymoma subsets including increased expression of SOX9[41]. SOX9 expression was positively correlated with multiple other genes includingPOU2F3and expression of TRPM5 and KIT[41]. These studies reveal that thymomas show heterogenous molecular alterations that can be corralled into relatively discrete groups. A morpho-molecular classification of thymomas will likely replace the traditional histomorphologic classification systems sometime in the future. This welcomed modification will simplify and improve the classification and prognostication of these lesions. However, at present, no recurrent clinically actionable mutations have been identified, and molecularly guided treatment decisions remain elusive.

Figure 3. Numbers of non-synonymous mutations in atypical (type B3) thymomas. A bar plot of next-generation sequencing data is shown from 47 atypical thymomas (unpublished data) using the AmpliSeq Cancer Hotspot Panel v2 (Illumina, product number 20019161) using paired-end 2 x 300 read format with an average 500X depth per sample. The bar plot demonstrates a low number of non-synonymous mutations, with the majority of samples showing only two or fewer mutations.
Molecular landscape of thymic carcinoma
Thymic carcinomas represent extremely rare cancers comprising approximately 15%-20% of all TETs. These are separated from their more indolent counterparts based on the presence of aggressive histological features including overt nuclear atypia, necrosis, infiltrative architecture, and loss of the normal organotypical features of the thymus found in thymomas[1,3]. Thymic carcinomas pose a challenge for diagnosis due to a harrowing degree of heterogeneity in their morphology; at least 12 different subtypes of thymic carcinoma have been described to date [Table 2]. Due to their incredibly rare nature, these tumors are difficult to study, and management of these patients may prove difficult in the absence of established molecular targets or consensus treatment guidelines. Limited data exist on the molecular underpinnings of the majority of these tumor types, with only few cases studied to date. In general, thymic carcinomas tend to show increased tumor mutational burdens, multiple copy number alterations, and varied, non-recurrent,somatic mutations[37]. The most prevalent of these tumors is squamous cell carcinoma and variants thereof.
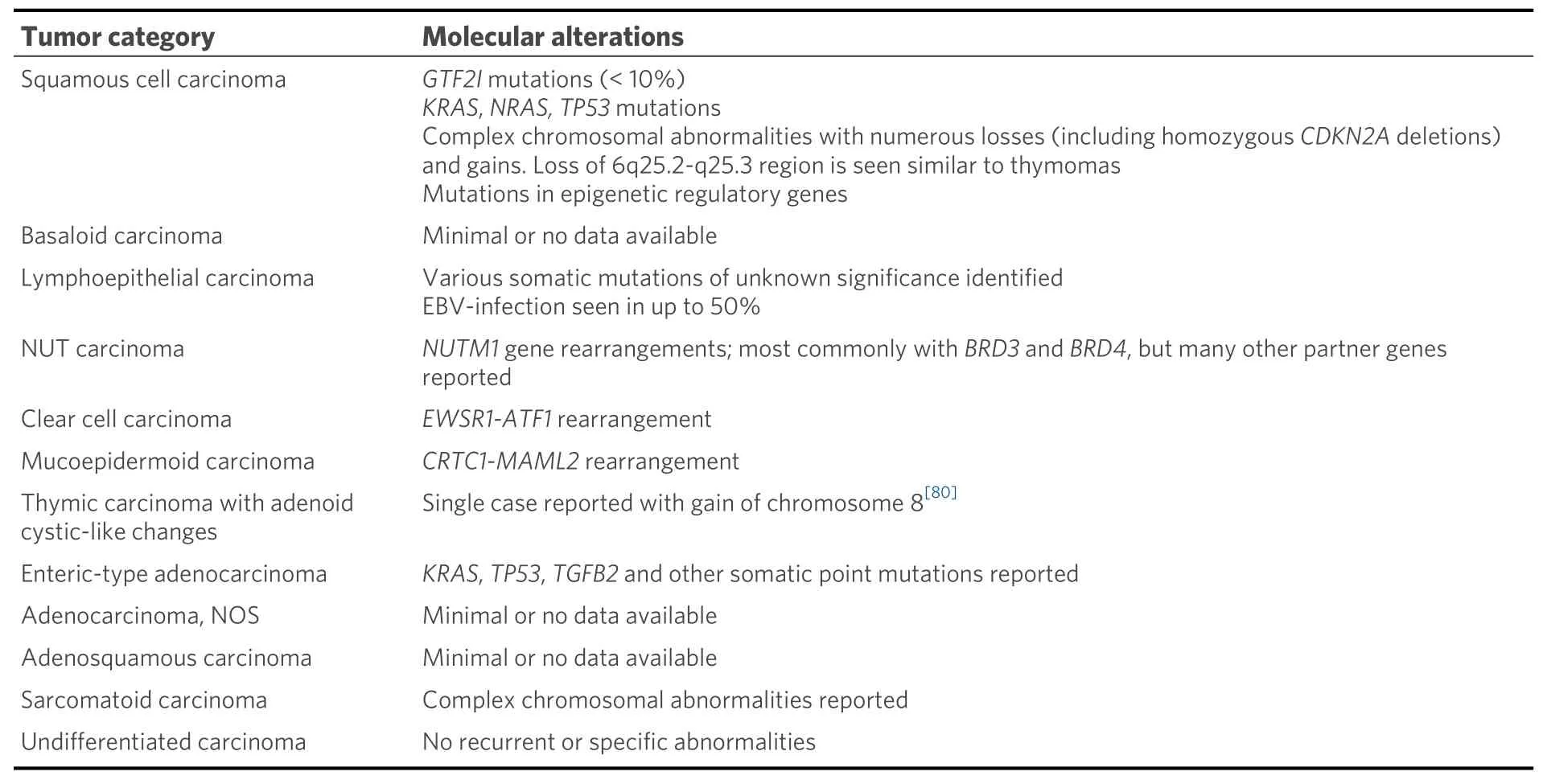
Table 2. Current WHO classification of thymic carcinoma and associated molecular alterations

?
Molecular studies regarding many other types of thymic carcinomas are extremely limited. Many subtypes such as basaloid squamous cell carcinoma, adenosquamous carcinoma, gland forming adenocarcinomas of the thymus, and sarcomatoid carcinomas have either only scattered reports or no testing reported[55-59].Interestingly, though, rare cases of primary thymic salivary gland tumors appear to show similar molecular alterations to their salivary gland counterparts includingEWSR1-ATF1fusion in clear cell hyalinizing carcinoma andCRTC1-MAML2fusion in mucoepidermoid carcinoma[60-62]. Finally, although rare, thoracic NUT carcinoma has been relatively well molecularly characterized and is known to harbor a characteristicBRD4-NUTM1fusion gene in approximately 75% of cases[63-67]. Alternative fusion partners includeBRD3(15% of cases), as well as other less common fusion partners such asNSD3,ZNF532, andZNF592(<5% of cases)[66-70].
CONCLUSION
It has only been in the past few decades that the molecular landscape of thymic epithelial neoplasms has begun to be defined. These tumors tend to show a heterogenous complement of molecular alterations,although multi-omic analyses have shown that they can be stratified into different categories based on some recurrent molecular alterations. Of interest are the losses documented at 6q25.2-q25.3 that are found both in thymic carcinoma as well as all subtypes of thymoma, suggesting that these tumors may, in some cases,evolve from pre-existing lower grade lesions; this is corroborated by the fact that thymic carcinomas may rarely be seen arising from thymomas of various subtypes, particularly in combination with atypical (WHO type B3) thymomas[71]. Despite this, many thymic carcinomas arise in the absence of a pre-existing thymoma, and the presence of significant differences in the documented underlying molecular alterations between the two indicates that thymic epithelial cells may arrive at malignancy through more than one molecular pathway. Clinical management and staging of thymomas can be difficult and should be approached in a multidisciplinary setting[72]. The discovery of recurrent alterations including point mutations, fusions, and characteristic micro RNA expressions indicate that these tumors may at some point be amenable to targeted therapies as new treatments are developed. A recent clinical trial (REMORA trial)demonstrated some efficacy with Lenvatinib in patients with advanced thymic carcinoma[73]. Some cases of thymic epithelial neoplasms have also shown alterations that modify cellular signaling pathways such as RAS and mTOR signaling and appear to be amenable to treatments directed at these pathways[74]. Recent studies using immunotherapy have shown conflicting results in aggressive thymic tumors, and additional studies are likely required to identify best use scenarios[75,76]. For now, molecular alterations such asGTF2Imutations orMAML2,EWSR1, andNUTrearrangements, are most valuable in a diagnostic capacity,although rare mutations, such asKITmutations, may be targeted with specific therapies. Future studies will seek to continue delineating the molecular characteristics of these tumors with the goal of eventually arriving at more precise morpho-molecular classifications that can be used to better guide prognosis,treatment and management of patients with TETs. In current clinical practice, patients with these tumors may benefit from advanced sequencing studies, particularly patients with thymic carcinomas, as the findings can inform the diagnosis and rarely help guide treatment if a clinically actionable mutation is detected.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study and performed data analysis and interpretation: Suster DI, Basu MK, Mackinnon AC
Availability of data and materials
Data source: Data included in this manuscript is available upon reasonable request of the corresponding author. Somatic point mutations across theGTF2Igene were obtained from St. Jude Cloud[77-79](https://www.stjude.cloud).
Financial support and sponsorship
None.
Conflicts of Interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2022.
杂志排行

Journal of Cancer Metastasis and Treatment的其它文章
- Anti-angiogenic drugs in cancer therapeutics: a review of the latest preclinical and clinical studies of anti-angiogenic agents with anticancer potential
- Tumors due to chronic exposure to benzene and biomarkers of exposure
- Lymph node metastasis from non-melanoma skin cancer
- Sensitivity of MCF-7 mammosphere CSCs to neutron radiation
- AUTHOR INSTRUCTIONS