随机对照临床试验设计要点和规范
2022-05-07王瑞平肇辉李斌
王瑞平 肇辉 李斌
摘 要 随机对照临床试验(randomized controlled clinical trial, RCT)是获取高级别循证医学证据的重要研究类别。为提高RCT研究的质量,研究者应在前期根据研究目的规范开展RCT研究设计,并从患者招募、样本量、随机化分组、盲法设置、干预措施实施、疗效评估、质量控制和统计学分析等方面进行综合考量。本文介绍了临床研究PICO(patients, intervention, comparisons, outcomes)原则和随机对照试验报告统一标准(consolidated standards of reporting trials, CONSORT)声明,详细阐述了RCT设计要点,为医务人员开展规范的临床研究设计提供参考。
关键词 随机对照临床试验 临床试验 研究设计 规范
中图分类号:R-3 文献标志码:C 文章编号:1006-1533(2022)07-0072-06
引用本文 王瑞平, 肇晖, 李斌. 随机对照临床试验设计要点和规范[J]. 上海医药, 2022, 43(7): 72-77.
Critical points and standards of randomized controlled clinical trial design
WANG Ruiping1, ZHAO Hui2, LI Bin1(1. Clinical Research & Innovation Center, Shanghai Skin Disease Hospital, Shanghai 200443, China; 2. Shanghai Pharmaceutical Profession Association, Shanghai 200003, China)
ABSTRACT Randomized controlled clinical trial (RCT) is critical for obtaining high-level evidence-based medical evidence. In order to improve the quality of RCT study, researchers should standardize the design of the RCT study according to the purpose of the study and comprehensively consideration should be made in terms of patient recruitment, sample size, randomization grouping, blind method setting, intervention implementation, curative effect evaluation, quality control and statistical analysis. This article introduces the PICO (patients, interventions, comparisons, outcomes) principles and the consolidated standards of reporting trials (CONSORT) statement and elaborates the critical points of RCT design so as to provide a reference for medical professionals to conduct standardized clinical research design.
KEy wORDS RCT; clinical trials; research design; standard
臨床试验是指以患者为研究对象,以个体为单位进行随机化分组,给予不同干预措施后,评价某种新药物或新疗法对疾病的疗效和安全性的研究[1]。临床试验研究是临床研究的重要组成部分,其核心内容为临床研究的设计(design)、测量(measurement)和评价(evaluation)[2]。近年来,随着医学学科发展和科研政策导向,临床研究工作越来越受到医务人员的重视与青睐。然而,许多医务人员在实际开展临床研究时遇到各种类型问题,归根到底是因前期的临床研究设计不规范所致。本文基于临床研究PICO(patients, intervention, comparisons, outcomes)原则和随机对照试验报告统一标准(consolidated standards of reporting trials, CONSORT)声明[3-5],从患者招募、样本量、分组、随机化、盲法设置、干预措施实施、疗效评估、质量控制和统计学分析等方面详细阐述随机对照临床试验(randomized controlled clinical trial, RCT)设计的要点,为医务人员今后开展规范的RCT研究设计提供参考。
1 RCT研究设计指导思想
规范的RCT研究设计应体现研究的代表性、真实性、可比性和显著性,进而才能做到研究的科学性、创新性和可行性。①“代表性”是保证研究结果科学性的基础和前提,若研究的代表性不强,不能被其他研究者重复,其研究结果将失去科学性。为提高RCT研究的代表性,研究者首先应根据研究目的,制定严格的研究对象诊断、纳入和排除标准,在选择研究对象时尽可能采取“随机化”抽样,使样本的具备良好代表性;其次,研究者应管理好入选的研究对象,与其保持紧密沟通,提高其依从性,并定期随访,降低失访率。②“真实性”是反映客观事物的正确程度,是科学性的核心要素。在RCT研究设计时,应全面考虑研究过程各环节的真实性,即如何真实无偏地采集和记录研究对象的人口学特征、身体测量指标、症状体征、临床表现、治疗效果等信息,如何平衡多中心RCT研究中不同中心的仪器测量、实验室检测一致性问题等。这些均须研究者认真思考,做到研究全过程预防和控制选择性偏倚、信息偏倚和混杂偏倚。③“可比性”是科学性的表现。事物之间有比较才有鉴别,在RCT临床研究中一定要设置对照组,同时在研究过程中要重视试验组和对照对象以及同一种对象之间在纳入和排除标准、数据采集、实验室检测、疗效评价等方面的可比性。④“显著性”是科学性的条件,经统计学检验得出显著性差异的结果才能体现研究的科学性。RCT研究设计时,研究者须在样本量计算和统计学分析部分体现“显著性”,通常以检验水准α代表“显著性”,α的常规取值为0.05或0.01,研究者可以根据具体情况灵活应用。
2 RCT研究设计的内容和要点
2.1 研究对象选择
研究对象是临床研究的灵魂,也是决定临床研究成败的关键因素。选择研究对象时,研究者应注意研究对象的选择标准、代表性、依从性和伦理符合性等问题。首先,RCT研究应根据研究目标确定研究对象的诊断标准,一般依据教科书、临床诊疗指南和规范制定,或依据科学共同体制定的标准制定。其次,研究者须制定研究的纳入标准和排除标准,要强调的是:排除标准不是纳入标准的互斥条件,而是在研究对象符合纳入标准的基础上具有一些特殊情况时应排除的条件,比如妊娠、治疗禁忌证等,故实际入组人群=符合诊断和纳入标准人群-符合纳入与排除标准人群。最后,对于一些少见病或因纳入标准严格导致研究对象来源困难时,研究者应权衡利弊,制定合适的标准,既保证研究的科学性,又照顾研究的实际可操作性。
2.2 样本量
样本量估算是RCT研究设计的重要内容之一。样本量过小不能保证得研究结论的可靠性;而样本量过大则会造成不必要的人力、物力和财力等浪费,同时增加研究难度。在样本量估算时,须明确几个参数:①Ⅰ类错误概率α,一般取0.01或0.05,α越小样本量越大。②把握度β,一般取0.1或0.2,β越小样本量越大。③允许误差δ,δ越小样本量越大。④干预的有效率(P1和P0)或疗效评价结果均值差值(D)和标准差(SD),P1和P0的差异越大,样本量越小;SD与D的比值越大,样本量越大。研究者可以根据实际情况,通过调整参数的选择确定合适的样本量。此外,由于临床研究的类型不同,研究的主要疗效结局指标不同,故样本量计算公式也不相同。样本量计算问题将在后续文章中以专题形式讲解,本文仅举例说明两组平行设计的临床试验研究的样本量估算[6]。

根据上述计算,得出每组需要23例研究对象,考虑10%脱落率,则每组须招募研究对象26例,两组共计52例。


根据上述计算,每组需要48例研究对象,考虑20%脱落率,每组须招募研究对象60例,两组共计120例。
2.3 设置对照组
上述“可比性”指导思想中已指出,两事物之间有比较才能鉴别,故“比较”为各种科学研究的基本方法。临床试验研究中,研究者可以根据研究目的和设计,选择随机对照、自身对照、交叉对照、非随机对照和历史对照。
随机对照是目前科学性最好、论证强度最高的一种对照方式,是指将研究对象按照不同的随机分配方案分为试验组和对照组,试验组给予待研究的干预因素,对照组给予现有的治疗措施、标准疗法或安慰剂。须注意的是,在随机对照中选择安慰剂对照或空白对照时,应注意伦理学问题。
自身对照是指以受试者本身作为对照,可以是受试者本身治疗前后对比,也可以是选择同一个受试者的不同受试部位进行同期对照(如皮肤、眼睛、口腔等)。对于肿瘤等慢性无自愈倾向的疾病,可以选自身前后对照进行疗效评估,但对于有自愈倾向的疾病(如上呼吸道感染、轻症肺炎等),不建议用自身对照。
交叉对照是指將研究对象随机分为试验组和对照组,整个研究包括2个阶段,第1阶段为试验组的受试者在第2阶段作为对照组,第1阶段为对照组的受试者在第2阶段作为试验组。须注意的是,交叉对照设计应在第1阶段结束后和第2阶段开始前设置间歇期(即洗脱期,一般不超过2周),同时在第2阶段开始前,试验组和对照组的基本情况应与第一阶段开始时完全一致(不能脱落病例),否则无法实施。
非随机对照是指研究对象未能随机分组的情况。由于未进行随机化分组,两组受试者在人口学特征、疾病严重程度等方面都可能存在差异,影响研究结果的评价,故不推荐使用。
历史对照是指在临床试验中仅设置试验组,而将以往治疗的一组同类疾病患者作为对照组进行比较。历史对照因未进行随机分组,同样具有一定的局限性。
2.4 随机化方案
RCT研究中,随机化分组是保证试验组和对照组除干预因素以外,其他因素在两组之间均衡可比的重要措施,也是控制研究偏倚的重要方法。临床研究中,常用的随机化方法包括:简单随机化、简单排序随机化、系统随机化、分层随机化、区组随机化、整群随机化、动态随机化和中央随机化系统。本文仅介绍常用的简单随机化、简单排序随机化和区组随机化3种分组方案。
简单随机化分组方案适合于小规模研究的随机化分组,通常是为获得期望的统计把握度则对患者的数量(组间分配比例)无特殊要求,对随机化序列不强加任何限制的随机化过程。这种方法操作简单,但会存在分组后组间样本量不等的局限性。
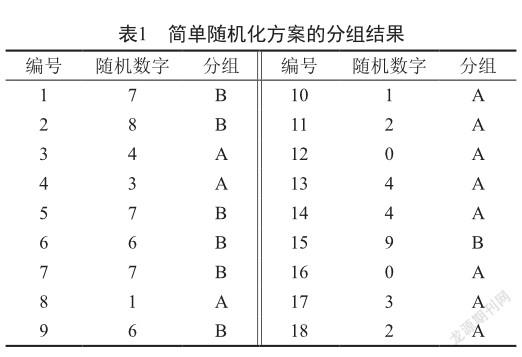
例如一项临床研究拟将18例患者进行随机分组为2组,采用简单随机化分组方案,从图1随机数字表[2]中第4行第1列开始选取18个1位数的随机数字,然后按照“0~4为A组,5~9为B组”规定即可将18例患者随机分为两组,其分组结果如表1所示,A组11例,B组7例。
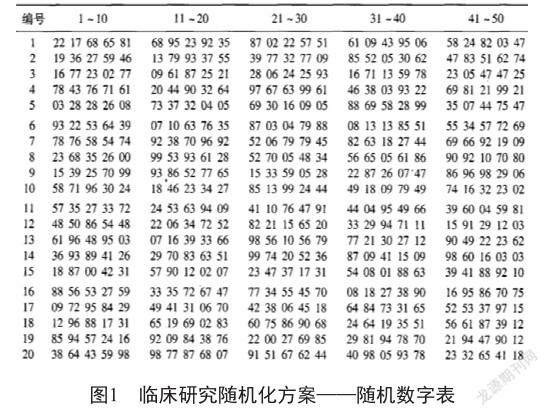
为解决简单随机化分组方案导致的组间样本例数不相等的问题,可采用简单排序随机化分组方案。该方案通过选取大小不等的随机数字进行排序,可保证各组例数相等,提高检验效能。同样以上述研究为例,拟将18例患者随机分为2组,要求两组样本量相等。采用简单排序随机化分组方案,从图1随机数字表中第七行第一列开始选取18个大小不等的2位数随机数字,如遇到相同随机数字则舍弃(本案例中为92和79),共得到18个随机数字,从小到大排序,规定排序序号1~9为A组,10~18为B组。如表2所示,18例患者随机分为2组,其中A组9例,B组9例。
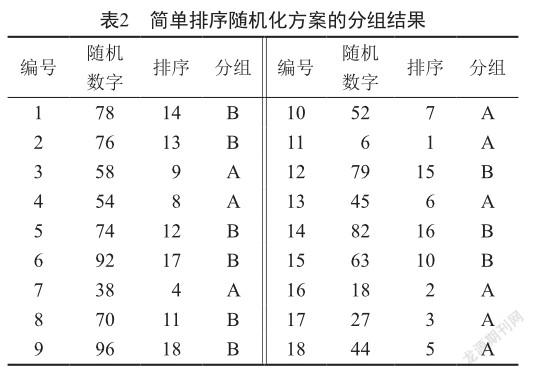
尽管通过简单排序随机化法可以将研究对象随机分为病例数一样的两组,但如果研究者入组的顺序受没有特征的影响(如经济状况、疾病严重程度等),采用简单排序随机化法将可能导致两组之间不均衡,研究对象之间不可比。如表2中的1~6号病例,随机化分组后,A组2例,B组4例,如果1~6号病例恰巧全部是重症病例,就会出现A组重症病例少,B组重症病例多的情况,影响疗效评估。为解决这个问题,可采用区组随机化分组方案进行分组。
区组随机化分组方案是先根据患者的某些特征(年龄/疾病严重程度等)进行排序,并划分为相同或不同间距的区组,然后在区组内应用简单排序随机化法方案进行分组,保证各组例数相等,提高组间均衡性,改善检验效能。同样以上述研究为例,拟将18例患者随机分为2组,要求两组样本量相等,同时考虑排除经济状况可能的影响。采用区组随机化分组方案,从图1随机数字表中第7行第1列开始选取18个大小不等的2位数随机数字,如遇到相同随机数字舍弃(本案例中为92和79),共得到18个随机数字。然后,将18个受试者编号换分为区组长度为4/6/4/4的4个区组(表3),每个区组内部按照简单排序随机化方案进行分组,可以将18个研究对象分为样本量相等的两组,同时也排除了经济情况的潜在影响。

RCT研究中,无论采取何种随机化方法以及研究方案是否设盲,为保证随机分配方案在执行过程中不受人为因素干扰,须采取随机化分配隐藏。随机化分配隐藏是指采取某些技术措施使参与研究的所有人员,包括研究人员、医生与研究对象均不知道随机化分配的顺序,常用的方法为编号的、不透明密封信封或药品容器。有条件的情况下可以使用中央随机化系统。研究者要注意的是,分配隐藏和盲法的作用不同,前者主要控制选择性偏倚,后者除控制选择性偏倚外,还可以控制信息偏倚。
2.5 盲法
RCT研究中,为避免研究人员、研究对象或统计分析人员等的主观心理作用造成的不真实结果,应在临床研究过程中使用盲法。常用的盲法包括单盲、双盲和三盲。研究者应根据研究的设计、干预措施的属性等方面综合考虑,合理选择盲法设置。
单盲是指受试者不清楚给予措施的性质,不知道自己被分配在试验组或对照组,而医生或研究者知道受试者分组的情况。
双盲是指受试者和研究人员(医生)均不知道受试者的具体分组情况,仅研究制定的人员知道受试者分组的情况。
三盲是指受试者、研究人员和统计分析人员均不知道受试者分组情况,仅研究者委托人员掌握随机分组号,直至试验结束,得到统计分析结果,统计学分析报告初稿撰写完成后才揭晓的情况。
2.6 研究因素
临床试验研究中,明确细化研究因素的衡量标准是确定研究因素的基本原则。应制定细致、全面、可行的标准明确研究因素与研究对象接触、暴露的方式和剂量等,保证所有研究对象接触或暴露于同质的研究因素,相互可比,不引入偏倚。
临床试验中,研究因素为药物、非药物治疗措施或其他治疗方案等干预措施。首先,研究者要明确干预措施的具体内容,给出明确详细的定义或规定。例如,干预措施为药物时,应给出药物通用名、商品名、生产厂家、批号;若使用安慰剂,须注明制备方法、安慰剂材料和剂量、外观形状等内容。其次,要给出干预措施的具体操作方法。例如,开展针刺干预女性压力性尿失禁临床研究时,研究因素部分要明确针刺的穴位、进针方式(是否捻转)、进针深度、留针时间、每周治疗频次、整体疗程等信息。
既往开展科研项目评阅时,研究者往往对研究因素撰写的重视程度不够,多数情况下研究者选择概括性描述,内容不够详实,影响专家对研究项目的质量评价。研究者须注意,RCT研究中,“研究因素”是核心内容,是整个临床试验的灵魂,因此一定要重视临床试验的研究因素,详细描述试验组和对照组研究对象所接受的干预措施的每一个细节内容,做到课题组外的其他人员根据描述可以实施完全一样的干预措施,保证研究因素的可复制性。
2.7 疗效评价指标
临床研究疗效指标的选择应把握其真实性和可靠性。真实性即要重视灵敏度和特异度,可靠性即要重点考虑指标的可重复性。RCT研究在疗效评估指标选择时,首先应优先选择真实性和可靠性均好的指标,提高研究效果评估的证据等级;其次,疗效评估指标一定要区分主要与次要,主要疗效评估指标一般只设置1个,用于临床研究疗效或安全性评价,同时也是计算样本量的参考指标;再次,疗效指标选择时还应该重视指标的科学性,指标不宜过多,应与课题组或研究团队的人力、物力相匹配,与实验室的检测能力和课题经费匹配;最后,除疗效评估指标外,研究者可以适当添加心理学、社会学和行为学指标,增加评价指标的丰度,但不宜过多。关于临床研究疗效评价指标更详细内容,可参考发表于《上海医院》杂志“临床研究规范”栏目的第2期内容“临床研究规范设计PICO原则”[7]。
2.8 质量控制
为保障临床试验研究的顺利开展,须在研究设计、项目启动、项目实施和数据统计分析等阶段开展全流程的质量控制。临床研究质量控制的核心内容是采取措施来避免或降低研究过程中可能会出现的偏倚,即研究者通过临床研究所取得的结果与真实的客观结果之间的系统误差,包括选择性偏倚(选择的研究对象不能代表目标人群)、信息偏倚(收集资料和测量指标的数据与信息不准确)和混杂偏倚(混杂因素的存在导致偏倚)。对于偏倚的定义、属性、分类及控制策略,后续将设置专题详细讲解,或参考《流行病学》等教科书自行学习。
2.9 统计学分析
临床研究特别是RCT的统计学分析首先要考虑数据集,统计分析集的选择是否正确将直接影响分析结果的可靠性。临床研究数据分析一般遵从意向性分析(intention to treat, ITT)原则。ITT原则是指主要分析应包括所有随机化的受试者,按其所分到的组别进行随访、评价和分析而不管其是否依从计划完成试验过程。ITT原则保证了原始的随机化分组,可避免由于破坏随机化而造成的偏倚。然而,在临床研究实践中,由于可能存在受试者脱落、改变治疗方案等情况,ITT原则贯彻困难。因此,临床研究数据一般按照ITT原则将数据集分为全分析集(full analysis set, FAS)和符合方案集(per protocol set, PPS)及安全集(safety set, SS)。其中FAS是指盡可能按照ITT原则,所有随机化的受试者以合理的方法尽可能少地排除受试者(排除不符合纳入与排除标准的入组者、未服药者、无任何数据者),部分受试者由于退出或剔除导致的数据缺失,可以通过末次观察值结转法(last observation carried forward, LOCF)进行数据填补并在数据统计分析中说明。PPS是FAS的一个子集,是更加符合研究方案的受试者数据集合,一般由完成了预先确定的治疗量、主要变量可测定、无重大方案违背的受试者组成。SS应包括所有随机化后至少接受一次治疗的受试者,用于安全性分析。
数据统计分析应优先使用FAS,特别是对于采用优效性设计的临床研究,应用FAS的分析结果更加保守和稳健。在统计学分析中,应明确4个方面的内容:①进行统计学分析的软件,一般为SAS、Epi info、SPSS和R软件等;②统计学描述;③统计学推断;④检验水准,一般设置α为0.05或0.01,并明确是单侧检验还是双侧检验。统计学分析这部分的详细内容请参考《上海医院》杂志“临床研究规范”栏目的第1期内容[8-10]。
3 RCT研究设计注意事项
开展临床研究设计时,研究者应按照上述第2部分中的9个方面进行考虑并执行,由此可基本保证方案设计的完整性、科学性和规范性。此外,研究者在开展临床研究设计时还要注意以下几个方面的问题:①在临床研究设计阶段,建议研究者邀请流行病学和统计学专家参与研究方案讨论,提高方案的规范性和可操作性。②临床研究设计要遵从PICO原则,并在“代表性”“真实性”“可比性”和“显著性”的中心思想指导下开展。③样本量估算时,要给出样本量计算公式中主要疗效指标参考值的来源,建议优先选择课题组的预实验结果,其次是通过类似研究的参考文献获取。但须强调的是,试验组和对照组的参考值要来源于同一篇文献。此外,样本量计算时选择的主要疗效指标应与研究方案的“疗效评价指标”中的主要疗效指标一致。④选择对照时,优先使用同期平行对照,便于随机化实施,提高研究对象的可比性进而提升研究证据循证医学等级。⑤无论临床研究选择开放性试验还是设置盲法,均要设置随机化分配隐藏,保障随机分配方案在执行过程中不受人为因素干扰。⑥研究者应避免对“单盲”的错误认识,“单盲”是指仅受试者不知道研究分组的情况,而评估人员不清楚受试者分组的情况不是单盲,是开放性试验。⑦研究因素是临床研究的核心内容,研究者要详细描述试验组和对照组研究对象所接受的干预措施的每个细节,保证“研究因素”的可复制性。⑧临床研究一般仅设置1个主要疗效评价指标,研究者应避免设置多个主要疗效评价指标,以防多个指标最终的指向性不一致,导致临床试验的结果无法给出评价,同时要尽可能选择客观指标。⑨在描述统计学分析部分时,要注意区分定量变量和定性变量在统计学描述和统计学推断的选择指标不同;此外,要注意P值结果解读的规范性,一般将“P<0.05(或0.01)”描述为差异有统计学意义,不可写成“差异有统计学显著性”;其次,也不能写成“P<0.05为差异有统计学意义,P<0.01为差异有显著性的统计学意义”,两者之间没有统计学差异的递进关系,研究者须避免。
参考文献
[1] Kakkar AK, Padhy BM, Sarangi SC, et al. Methodological characteristics of clinical trials: impact of mandatory trial registration[J]. J Pharm Pharm Sci, 2019, 22(1): 131-141.
[2] 詹思延. 临床流行病学[M]. 2版. 北京: 人民卫生出版社, 2015.
[3] Schiavenato M, Chu F. PICO: what it is and what it is not[J]. Nurse Educ Pract, 2021, 56: 103194.
[4] Jüni P, Altman DG, Egger M. Systematic reviews in health care: assessing the quality of controlled clinical trials[J]. BMJ, 2001, 323(7303): 42-46.
[5] Schulz KF, Altman DG, Moher D, et al. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials[J]. BMJ, 2010, 340(3): 698-702.
[6] 邓伟, 贺佳. 临床试验设计与统计分析[M]. 北京: 人民卫生出版社, 2012.
[7] 王瑞平. 临床研究规范设计PICO原则[J]. 上海医药, 2022, 43(3): 67-72.
[8] 王瑞平, 李斌. 临床医学研究數据分类浅谈[J]. 上海医药, 2022, 43(1): 3-6.
[9] 王瑞平, 李斌. 临床医学研究数据统计分析思路概述[J].上海医药, 2022, 43(1): 7-9.
[10] 王瑞平, 李斌. 临床医学研究数据库的创建和质量控制要点[J]. 上海医药, 2022, 43(1): 10-14.
