准东煤燃烧过程中钙硅镁分子动力学研究
2022-04-18郭万贵吕为智
郭万贵,吕为智
(1.国家电力投资集团有限公司新疆能源化工有限责任公司,乌鲁木齐 830010;2.上海发电设备成套设计研究院有限责任公司,上海 200240)
新疆准东煤田是我国最新发现的预测储量达到3 900亿t的巨大露天煤矿,是目前我国最大的整装煤田,准东煤田将成为我国十分重要的能源接续区和战略性储备区[1]。在准东煤燃烧过程中,锅炉受热面极易出现严重结渣、沾污现象,这极大地限制了准东煤的应用[2]。高钙准东煤灰中碱土金属钙含量很高,在燃烧过程中易与煤灰中铝、镁、硅等矿物形成低温共熔物,达到熔融状态后析出,造成严重的结渣和沾污现象[3-4]。
Wang等[5]对某330 MW燃准东煤锅炉沿烟气流程进行取样,结果表明在850 ℃左右硫酸钠和硫酸钙明显冷凝沉积在换热面,在1 000 ℃后硫酸钙开始分解,释放出的钙与硅氧化物形成复合硅酸盐继续沉积在表面,引起严重的积灰和结渣问题。Li等[6]对比了准东煤与高熔点烟煤、呼伦贝尔褐煤燃烧过程中的灰沉积现象,发现相比其他2种煤,准东煤的灰沉积现象更严重,准东煤灰中Ca、Na含量较高,硅灰石和钙长石等物质会形成一种富含Ca、Mg、Fe、Si及Al且熔点在900~1 100 ℃的共晶混合物,这是导致准东煤结渣的主要原因。
钙镁黄长石(Ca2MgSi2O7)是准东煤灰在高温下的主要钙类矿物质之一,也是影响准东煤结渣的重要因素。研究钙镁黄长石的生成机理对进一步深入揭示准东煤结渣机理具有重要意义。分子动力学可考虑浓度和温度等因素,使计算过程更贴合实际。因此,笔者通过分子动力学方法从微观角度探讨准东煤灰中钙镁黄长石的生成机理,使用分子动力学模拟反应物间不同的物质的量比(以下简称配比)及不同反应温度下的吸附、扩散特性,进一步揭示不同工况对钙镁黄长石生成机理的影响,为抑制钙镁黄长石的形成提供理论依据。
1 计算模型和方法
1.1 计算模型
建立以CaO、MgO、SiO2和CaSiO3为主要物质的基础模型,采用无机晶体结构数据库(ICSD)[7]的晶胞作为初始晶胞,使用CASTEP软件包进行构型优化,计算所得各模型的晶体结构如图1所示。在计算过程中,考虑到第一性原理中的广义梯度近似(Generalized Gradient Approximation,GGA)中的Perdew-Burke-Ernzerhof(PBE)泛函计算可适用于绝大部分元素[7],其对于非金属、金属体系均适用,采用GGA-PBE算法描述电子交换相关作用。Energy cutoff设置为300 eV,几何构型优化的收敛标准如下:自洽场(SCF)收敛精度为1.0×10-5eV,最大力为0.03 eV/Å,最大位移为0.001 Å,K点取值为4×4×1。使用Ultrasoft的赝势进行处理。
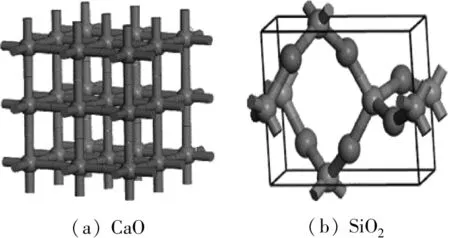
图1 各模型物质的几何构型Fig.1 Geometric configurations of various substances
计算所得CaO、SiO2、CaSiO3和MgO结构的晶格参数与实验中各模型物质的晶格参数对比如表1所示。由表1可知,计算结果与实验值具有较好的一致性。
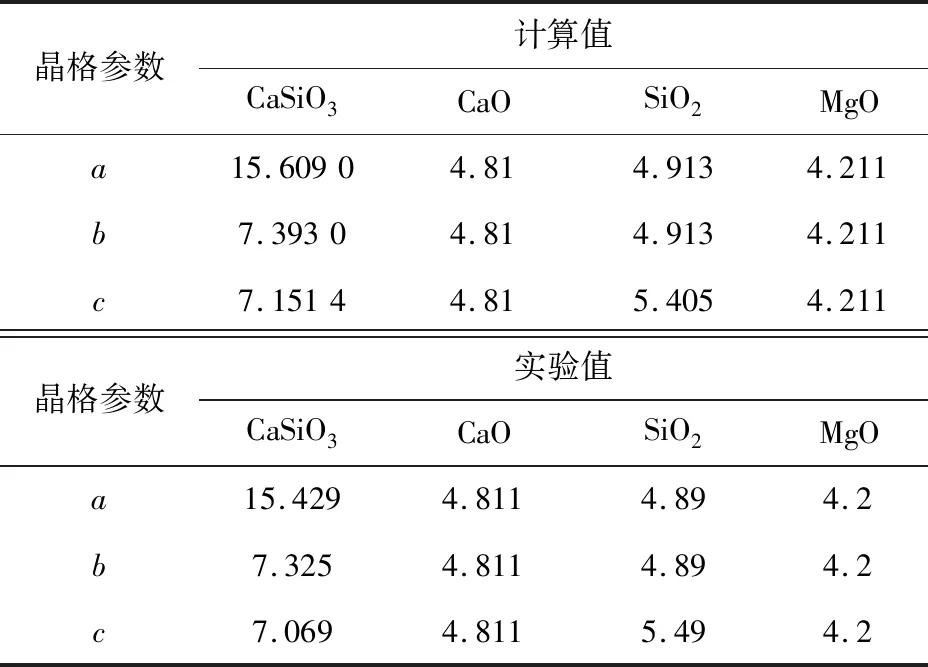
表1 各结构晶格参数计算值与实验值的对比Tab.1 Comparison of lattice parameters of various structures between calculated values and experimental values
CaO衍射峰暴露的晶面主要是(100)表面,CaSiO3衍射峰暴露的晶面主要是(001)表面[8]。同时,Hou等[9]的研究表明CaO稳定存在的表面是(100)表面。郑烨等[10]利用透射电子显微镜观察CaSiO3晶体的生成过程,其稳定存在的表面是(001)表面。在钙镁黄长石的形成过程中,首先是大量的CaO与SiO2反应,因此选取CaO(100)表面作为吸附基底,计算SiO2在CaO表面的结合、扩散特性;选取CaSiO3(001)表面作为吸附基底,计算MgO在CaSiO3表面的结合、扩散特性。
1.2 计算方法
基于分子动力学方法,分别对SiO2在CaO表面、MgO在CaSiO3表面的结合、扩散过程进行计算。考虑到COMPASS力场是基于第一性原理计算结果拟合的力场,其具有良好的普适性。此外,利用COMPASS力场对钙硅等固相体系、碳氢燃料等气相体系进行的分子动力学模拟也得到了比较理想的结果。笔者采用Discover软件进行模拟,采用COMPASS力场描述分子间的作用力,并采用Atom Based截断法计算非键原子间的范德华作用和库仑作用。由于表面吸附中起主要作用的是上两层原子,而深处的固体分子的影响可以忽略不计。因此,分别将CaO(100)表面CaSiO3(001)固体层中除上两层原子外全部固定,对其进行表面驰豫,并拓展成5×5的超胞模型作为基底,厚度为11.5 nm(力场的no-bond cut-off非键截断距离设置为9.5 Å),并在Z轴方向建立8 nm厚度的真空层,用以消除上层分子的影响。
最后,对不同温度、不同配比下SiO2在CaO(100)表面上的结合及扩散进行动力学分析。同理,对MgO在CaSiO3(001)表面上的结合及扩散进行动力学分析。所有模拟均采用NVT(保持原子数、体积和温度恒定)系综,且采用Benredsen热浴方法进行控温,弛豫时间为0.1 ps,时间步长为1.0 fs,模拟步数为200 000,总模拟时间为200.0 ps,每100步输出全部结构和运动轨迹,其中前100 ps使体系达到平衡,后100 ps用于进行各种性质分析。
2 结果与讨论
2.1 配比对CaO与SiO2结合特性的影响
钙镁黄长石生成机理的第一步是CaSiO3的形成,CaO与SiO2的配比对CaSiO3的形成有重要影响。因此,研究不同配比下SiO2在CaO表面的结合特性,从分子水平确定两组分在不同配比下的相互作用强弱,对控制硅灰石的形成具有重要意义。由于在1 100 ℃时会形成大量的钙镁黄长石,在此温度下建立CaO与SiO2的混合模型,图2为CaO与SiO2的配比r1为75∶25、70∶30、65∶35、60∶40、55∶45、50∶50、45∶55和40∶60时的结合形态图。
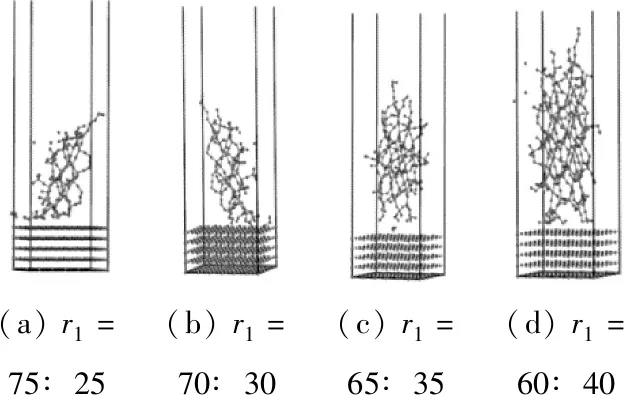
图2 CaO与SiO2混合模型在不同配比下的平衡结构Fig.2 Equilibrium structures of CaO and SiO2 mixed model at different ratios
不同配比下SiO2在CaO(100)表面的结合能如表2所示。结合能可以评估两组分间相互作用的强度。结合能若为负值,代表放热反应,其绝对值越大,体系之间越稳定;结合能若为正值,代表吸热反应,其数值越大,体系间越不稳定[11]。结合能的数学表达式如下:
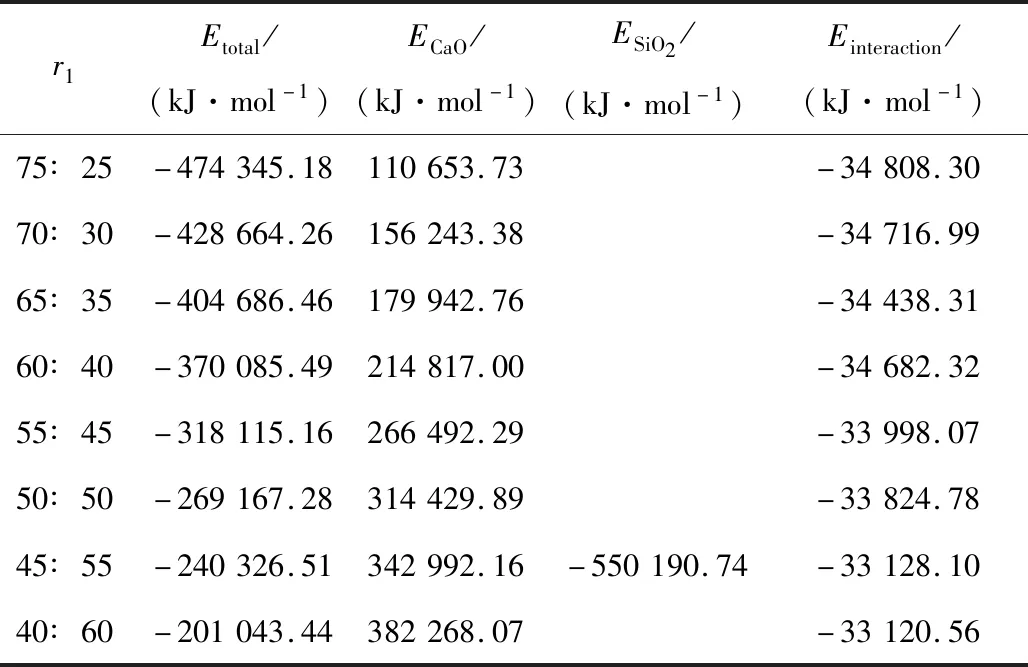
表2 不同配比下SiO2在CaO(100)表面的结合能Tab.2 Binding energy of SiO2 on CaO(100)surface at different ratios
Einteraction=Etotal-(ECaO+ESiO2)
(1)
式中:Einteraction为两组分间的结合能;Etotal为SiO2在CaO(100)表面结合达到平衡时的总能量;ECaO为基底CaO的能量;ESiO2为体系中SiO2的能量。
由表2可知,不同配比下SiO2在CaO(100)表面的结合能均为负值,为放热反应。当发生放热反应时,结合能的绝对值越大,表明SiO2在CaO(100)表面的黏附能力越强,越易生成CaSiO3。不同配比下SiO2在CaO(100)表面的结合能绝对值变化曲线如图3所示。由图3可知,在CaO与SiO2配比为75∶25时,两者的结合能绝对值最大,此时SiO2与CaO的相互作用力最强,最易形成CaSiO3;在配比为40∶60时,两者的结合能绝对值最小。随着配比的减小,SiO2在CaO(100)表面的结合能绝对值基本呈现下降趋势,但下降幅度不大。表明不同配比下CaO与SiO2的结合能有所不同,且当CaO物质的量占比越高时,结合能绝对值越大,SiO2在CaO(100)表面的黏附力越强,越易结合生成CaSiO3。

图3 不同配比下SiO2在CaO(100)表面结合能绝对值的变化Fig.3 Changes in the absolute value of binding energy of SiO2 on CaO(100)surface at different ratios
为进一步获得SiO2在CaO表面各原子间的相互作用机制,在上述分子动力学模拟的基础上进行径向分布函数(RDF)分析。径向分布函数又称关联函数g(r),是指以一个空间粒子为坐标,周围其他粒子在该空间的概率密度,代表物质的短程有序性以及电子相关性,可用来描述分子间的相互作用。其表达式为:
(2)
式中:r为原子间的键长距离;N′为原子总数量;V为体系的体积;n(rij)为j粒子围绕i粒子的个数;Δr为球壳的厚度。
由前文可得,在CaO与SiO2配比为75∶25时,两者的结合能绝对值最大,相互作用力最强,因此以该配比为例研究CaO与SiO2在原子层面的相互作用,如图4所示。图中SiO2分子中的Si、O原子分别用SiO2-Si、SiO2-O表示,CaO分子中的Ca、O原子则分别用CaO-Ca、CaO-O表示。
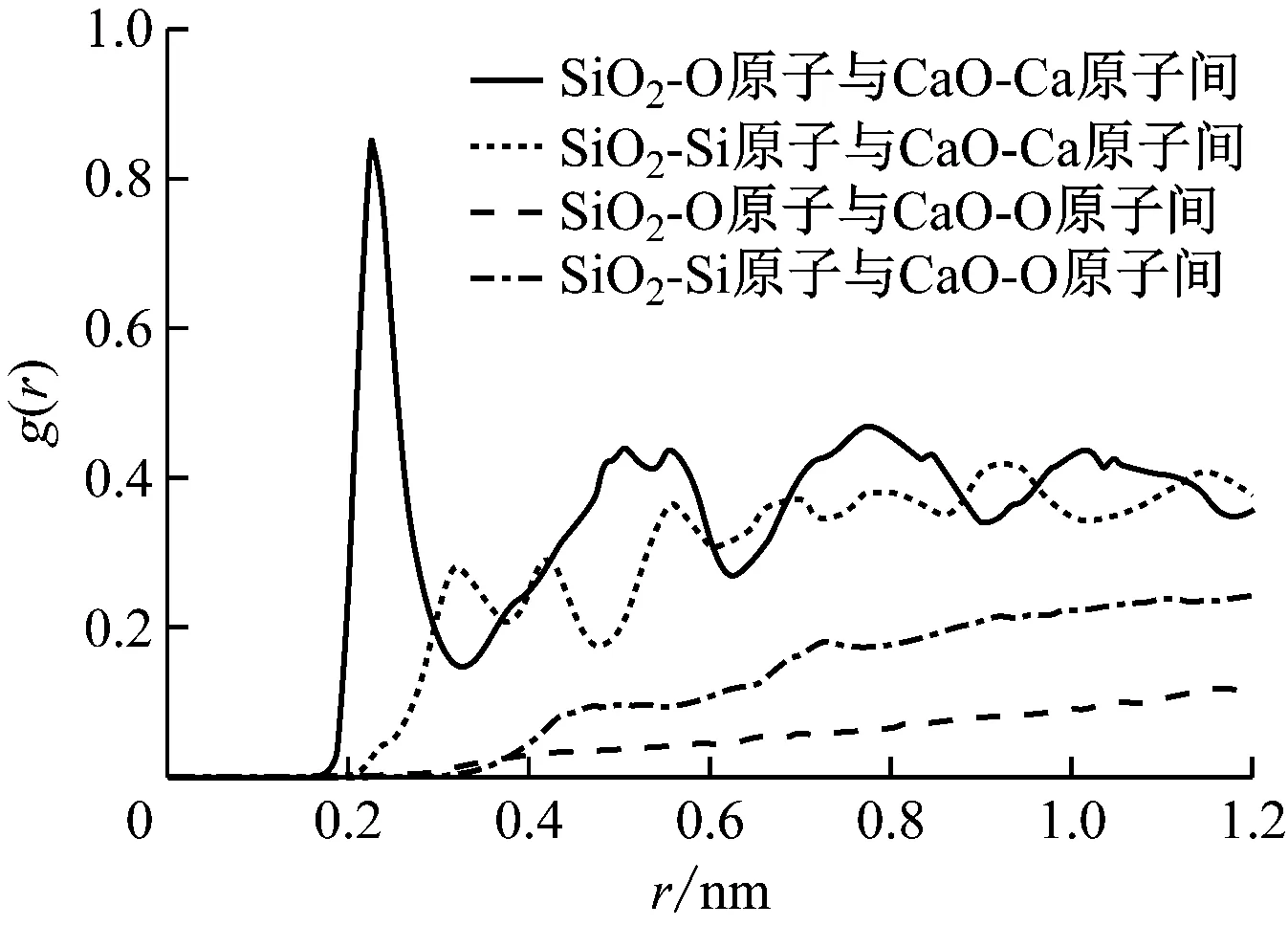
图4 75∶25配比下CaO与SiO2分子间的径向分布函数Fig.4 Radial distribution function between CaO and SiO2 molecules at the ratio of 75∶25
在径向分布函数图中,距离小于0.31 nm的峰主要是氢键作用和范德华作用,此时为化学吸附,表明有化学反应进行;大于0.31 nm的峰主要是范德华作用和库仑作用,但此时范德华作用很弱,界面间的相互作用为物理吸附[12]。由图4可知,SiO2-O原子与CaO-Ca原子间的g(r)、SiO2-Si原子与CaO-Ca原子间的g(r)在较长距离范围内均呈现良好的有序性,表明上述原子间的作用力较广,受化学键作用、范德华作用和库仑作用的共同影响。SiO2-Si原子与CaO-O原子间呈近程有序性,而距离Si原子较远的O原子分布无序化程度较高,说明这两种原子间存在较弱的物理吸附作用。SiO2-O原子与CaO-O原子间的g(r)基本呈现无序化状态,这两种原子间的相互作用力十分微弱。SiO2-O原子与CaO-Ca原子间的g(r)在0.23 nm处出现了高为0.86的峰值,表明这两种原子间受范德华作用,形成了稳定的化学键;在大于0.31 nm时曲线中包含多处明显的尖峰,且峰值明显小于近程位置的峰值,这是因为此时范德华作用较弱,SiO2-O原子与CaO-Ca原子间形成了物理吸附。SiO2-Si原子与CaO-Ca原子间的g(r)在0.3 nm左右出现了一个高为0.3的峰,且在中远程位置出现的峰值比该峰值高,说明SiO2-Si原子与CaO-Ca原子间存在较强的物理吸附作用。同时,对比SiO2-O原子与CaO-Ca原子间的g(r)以及SiO2-Si原子与CaO-Ca原子间的g(r)曲线,发现前者出现第一个峰的距离较短,且峰值较高,因此SiO2-O原子与CaO-Ca原子间的相互作用力相对更强,形成的化学键更稳定。综上可知,CaO与SiO2的作用力主要是SiO2-O原子与CaO-Ca原子间以及SiO2-Si原子与CaO-Ca原子间的相互作用为主,这也进一步揭示了前面CaO与SiO2的反应路径中是以Ca原子键合SiO2上的O原子、Ca原子键合SiO2上的Si原子这两种结合方式进行的原因。
同时,对不同配比下CaO-Ca原子与SiO2-O原子间以及CaO-Ca原子与SiO2-Si原子间的径向分布函数进行分析,如图5所示。由图5(a)可知,不同配比下SiO2-O原子与CaO-Ca原子间的径向分布函数各曲线均在0.25 nm处出现第一个明显的峰,但峰值有所不同。在CaO与SiO2的配比为75∶25时,第一个峰的峰值最高,其次是在配比为70∶30时第一个峰的峰值较高,且随着配比的减小,第一个峰的峰值也逐渐下降,表明CaO-Ca原子与SiO2-O原子间形成的Ca—O化学键强度减弱。由图5(b)可知,SiO2-Si原子与CaO-Ca原子间的径向分布函数曲线出现第一个峰的距离在0.3 nm左右,在CaO与SiO2的配比为75∶25和70∶30时的峰值较高,不同配比下各曲线中第一个峰的峰值随着配比的减小而下降。同时,配比为60∶40时的径向分布函数曲线在大于0.3 nm的中远程位置处的峰值明显升高,导致SiO2-Si原子与CaO-Ca原子间的物理吸附作用增强。由此可见,随着CaO物质的量占比的减少,两分子间起主要作用的SiO2-O原子与CaO-O原子间与SiO2-Si原子与CaO-Ca原子间的化学键强度减弱,因此二者的结合能也随之下降,而在60∶40配比时Ca—Si键的物理吸附作用增强,使得结合能略有升高。
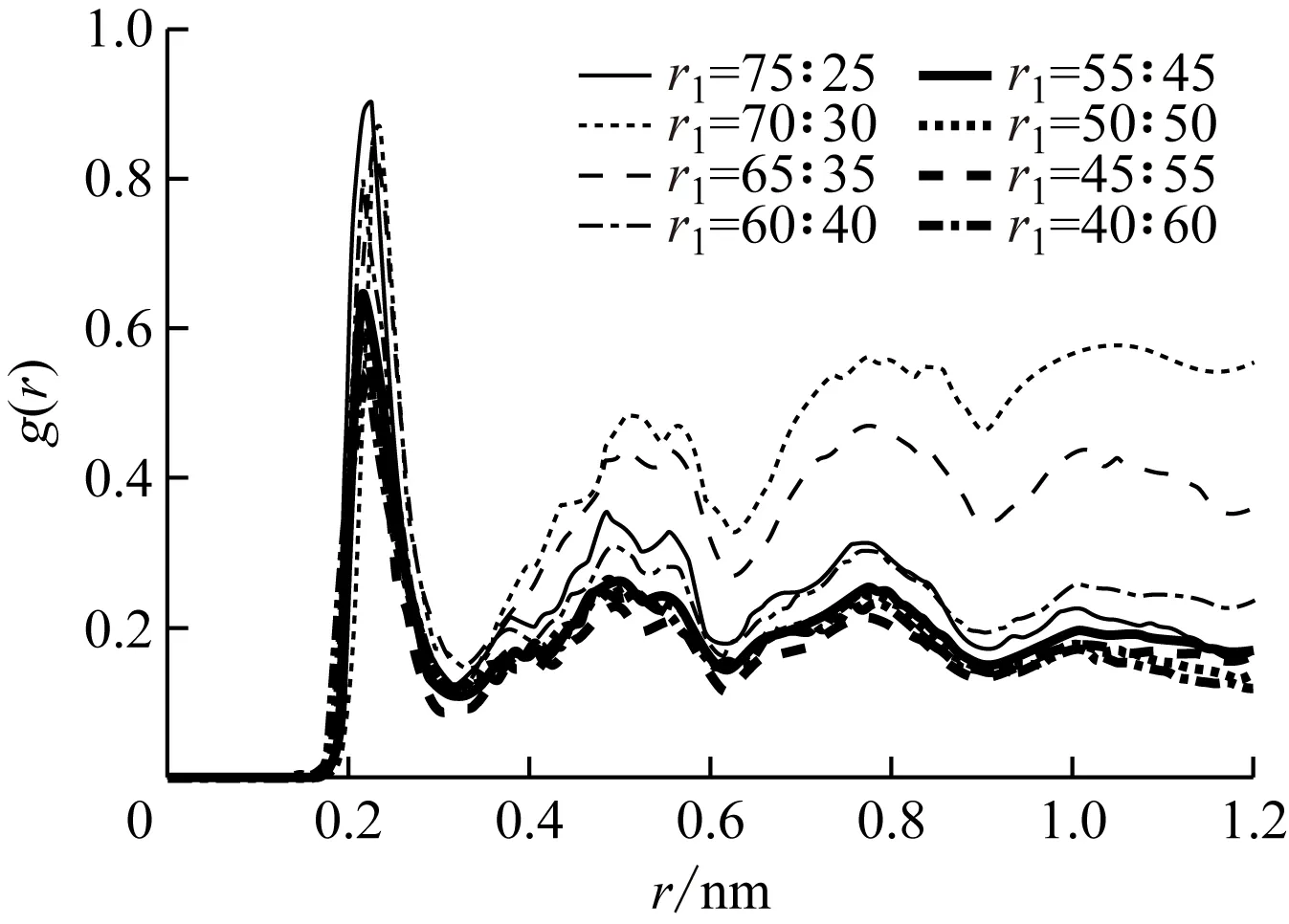
(a)SiO2-O原子与CaO-Ca原子间的径向分布函数
2.2 配比对CaSiO3与MgO结合特性的影响
钙镁黄长石形成的第二步是CaSiO3与MgO的反应。因此,笔者将研究不同配比下CaSiO3主要生长面(001)表面对MgO的结合特性。根据前期实验结果,在1 100 ℃时CaSiO3与MgO反应形成大量钙镁黄长石[13],因此在此温度下建立CaSiO3与MgO的混合模型,MgO的物质的量占比以5%的增幅从25%增至60%。图6为CaSiO3与MgO配比r2为75∶25、70∶30、65∶35、60∶40、55∶45、50∶50、45∶55和40∶60时的结合形态图。
图6 CaSiO3与MgO混合模型在不同配比下的平衡结构Fig.6 Equilibrium structures of CaSiO3 and MgO mixed model at different ratios
不同配比下MgO在CaSiO3主要生长面的结合能如表3所示。由表3可知,不同配比下MgO在CaSiO3(001)表面的结合能均为负值,是放热反应。当发生放热反应时,结合能的绝对值越大,体系则越稳定,相应MgO越易黏附在CaSiO3(001)表面,两者越易结合形成钙镁黄长石。CaSiO3与MgO的配比为60∶40(即3∶2)时,CaSiO3更易与MgO反应形成钙镁黄长石,而在配比为75∶25时,两者最不易结合。
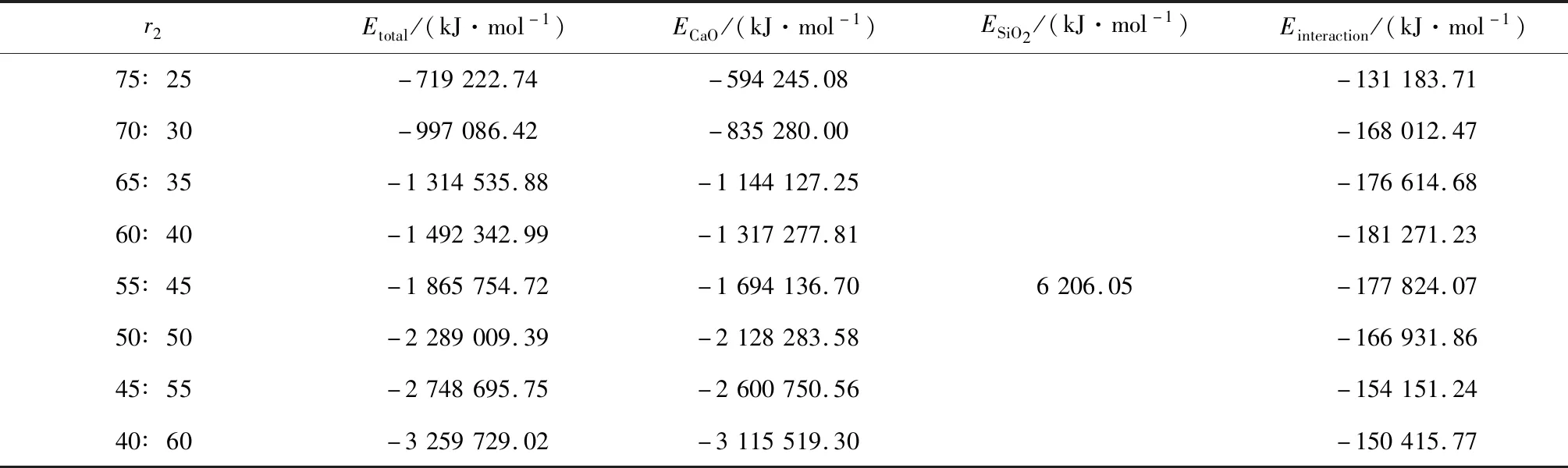
表3 不同配比下MgO在CaSiO3(001)表面的结合能Tab.3 Binding energy of MgO on CaSiO3(001)surface at different ratios
不同配比下MgO在CaSiO3(001)表面的结合能绝对值变化趋势如图7所示。由图7可知,随着MgO物质的量占比的增加,MgO在CaSiO3(001)表面上的结合能绝对值呈现先增加后下降的趋势。在CaSiO3与MgO的配比为60∶40时,两者结合能绝对值达到最大,为181 271.23 kJ/mol,表明MgO物质的量占比为40%时,两组分的相互作用最强;配比为65∶35和55∶45时CaSiO3与MgO的结合能绝对值相差不大,分别为176 614.68 kJ/mol和177 824.07 kJ/mol,略小于配比为60∶40时的结合能绝对值;配比为70∶30与50∶50时的结合能绝对值非常相近,分别为168 012.47 kJ/mol和166 931.86 kJ/mol,两者呈现对称式分布。
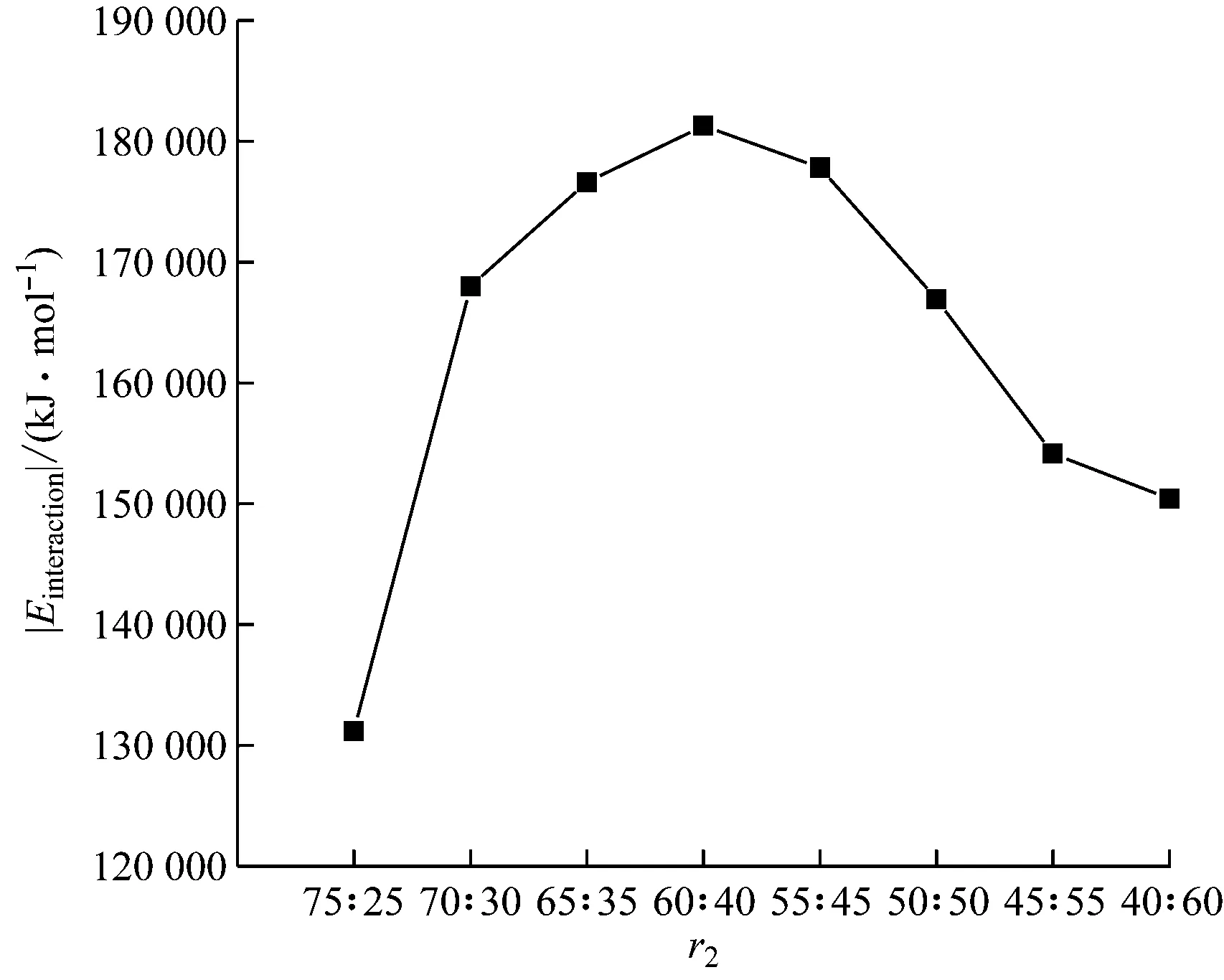
图7 不同配比下MgO在CaSiO3(001)表面结合能绝对值的变化Fig.7 Changes in the absolute value of binding energy of MgO on CaSiO3(001)surface at different ratios
CaSiO3分子中的Ca、Si和O原子分别用CaSiO3-Ca、CaSiO3-Si和CaSiO3-O表示,MgO分子中的Mg、O原子分别用MgO-Mg和MgO-O表示。以CaSiO3与MgO结合能最强时的配比60∶40为例,研究MgO分子中各原子对CaSiO3分子中各原子的相互作用,如图8所示。由图8可知,MgO-O原子与CaSiO3-Si原子间的g(r)以及MgO-Mg原子与CaSiO3-O原子间的g(r)在较长距离范围内均呈现良好的有序性,说明原子间的作用力较广,受化学键、范德华作用和库仑作用的共同影响。MgO-O原子与CaSiO3-Si原子间的g(r)在0.2~0.3 nm内出现了一个高为2.75的峰值,MgO-Mg原子与CaSiO3-O原子间的g(r)在0.25~0.35 nm内出现了一个高为0.75的峰值,表明MgO-O原子与CaSiO3-Si原子间以及MgO-Mg原子与CaSiO3-O原子间受范德华作用,存在强化学键。此外,MgO-O原子与CaSiO3-Si原子间出现第一个峰时的键长距离明显小于MgO-Mg原子与CaSiO3-O原子间的键长距离,且峰值较高,表明MgO-O原子与CaSiO3-Si原子间的相互作用更强。
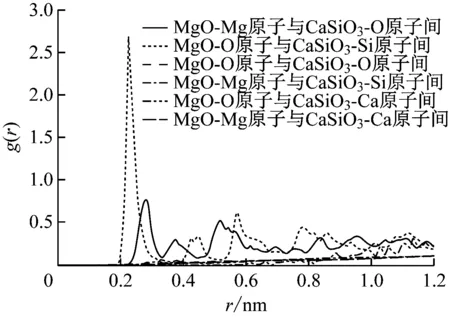
图8 60∶40配比下CaSiO3与MgO分子间的径向分布函数Fig.8 Radial distribution function between CaSiO3 and MgO molecules at the ratio of 60∶40
MgO-O原子与CaSiO3-Ca原子间的g(r)在大于0.6 nm处呈现微弱波峰,说明MgO-O原子与CaSiO3-Ca原子间受范德华作用和库仑作用的影响,存在较弱的物理吸附。MgO-O原子与CaSiO3-O原子间、MgO-Mg原子与CaSiO3-Ca原子间以及MgO-Mg原子与CaSiO3-Si原子间的g(r)基本呈现无序化状态,表明MgO-O原子与CaSiO3-O原子间以及MgO-Mg原子与CaSiO3-Ca原子、CaSiO3-Si原子间几乎不存在相互作用力。综上分析,CaSiO3分子与MgO分子间作用力的本质主要是MgO-O原子与CaSiO3-Si原子间以及MgO-Mg原子与CaSiO3-O原子间的相互作用,从而形成强的化学键。这一结论与钙镁黄长石的量子化学研究结果[12]吻合,钙镁黄长石形成的微观机理正是络合后的CaSiO3分子中的Si原子与O原子分别吸引MgO中的O原子和Mg原子,形成新的Si—O键与Mg—O键使体系达到平衡,趋于稳定结构后形成钙镁黄长石。
同时,分析不同配比下CaSiO3与MgO分子间的径向分布函数,从而探究起决定作用的MgO-O原子与CaSiO3-Si原子、CaSiO3-O原子间的相互作用力。由图9可知,在70∶30、65∶35、60∶40、55∶45和50∶50配比下,MgO-O原子与CaSiO3-Si原子间以及MgO-Mg原子与CaSiO3-O原子间出现第一个峰时的距离几乎没有明显差异,均存在强化学键作用,但峰值有所不同。在60∶40配比下,图9(a)和图9(b)出现第一个峰时的峰值最高,分别为2.75和0.75,表明此时MgO-O原子与CaSiO3-Si原子间以及MgO-Mg原子与CaSiO3-O原子间的相互作用力最强,因此两分子间的结合能绝对值亦最大。其次是在65∶35、55∶45配比下出现第一个峰时的峰值较高,且在近距离处的g(r)曲线非常相似,这与二者的结合能数值相近且仅次于60∶40配比下结合能的现象一致。在75∶25、45∶55和40∶60配比下的g(r)曲线在大于0.5 nm的较远位置处的波峰较强,此时主要受弱范德华作用和库仑作用,为物理吸附,且配比为45∶55和40∶60时的g(r)曲线在小于0.4 nm处几乎没有波峰出现,表明不存在强化学键和强范德华作用,因此相对其他配比下较强的化学吸附特性,这三者的结合能绝对值较小。

(a)MgO-O原子与CaSiO3-Si原子间的径向分布函数
2.3 温度对SiO2在CaO表面结合、扩散特性的影响
CaO与SiO2的结合能随着CaO物质的量占比的增加而增加,在CaO与SiO2的配比为75∶25时,两者的结合能绝对值最大。选择该配比下的模型研究不同温度下SiO2在CaO(100)表面的结合能,如表4所示。
由表4可知,随着温度的升高,SiO2在CaO(100)表面的结合能绝对值逐渐增加,但结合能的数值相差不大。这表明温度升高对SiO2在CaO(100)表面的结合有一定促进作用,但影响并不大。研究发现,不同的温度会对固体反应间的扩散特性产生重要影响[14]。因此,对SiO2在CaO(100)表面的分子扩散进行分析,得到其均方位移(MSD)曲线。均方位移可体现分子运动的稳定性,相同时间内的均方位移越大,表明分子间运动越剧烈[15]。不同温度下SiO2在CaO(100)表面的均方位移曲线如图10所示。

表4 不同温度下SiO2在CaO(100)表面的结合能Tab.4 Binding energy of SiO2 on CaO(100)surface at different temperatures
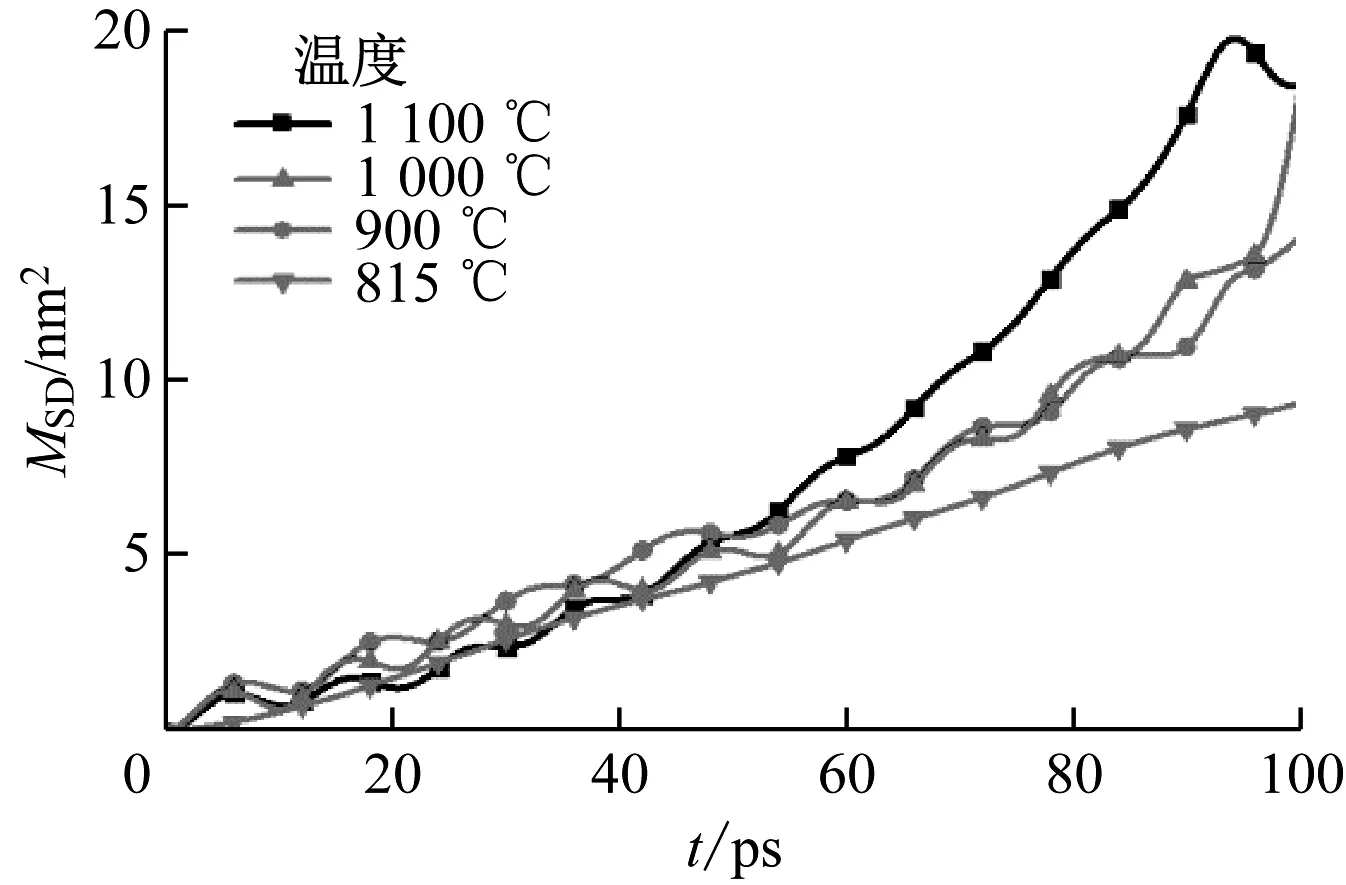
图10 不同温度下SiO2在CaO(100)表面的均方位移Fig.10 Mean square displacement of SiO2 on CaO(100)surface at different temperatures
由图10可知,当温度从815 ℃上升到1 100 ℃,SiO2在CaO(100)表面的均方位移随着时间的推移逐渐增大,这是因为温度升高使分子的内能增加,基于能量守恒,分子内能转化为分子动能,导致分子运动更加剧烈,在空间的迁移量增大。同时,对均方位移进行线性拟合,根据Einstein法得到扩散系数,其表达式为:
(3)
式中:ri(t)和ri(0)分别为分子i在t时刻及初始时刻的位置;N为分子数量。
通过MSD可以计算得到扩散系数D:
(4)
对均方位移进行线性拟合,求其斜率k,则扩散系数可简化为:
(5)
从而求得不同温度下SiO2在CaO表面的扩散系数,如表5所示。由表5可知,SiO2在CaO表面的扩散系数随温度的升高而增大,说明温度越高,SiO2分子越易扩散到CaO中。
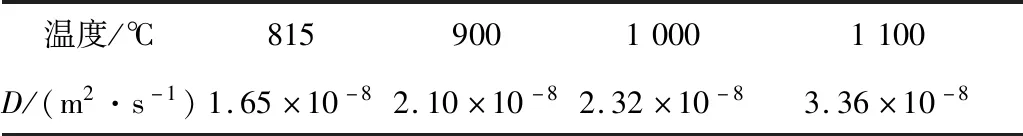
表5 不同温度下SiO2在CaO表面的扩散系数Tab.5 Diffusion coefficient of SiO2 on CaO surface at different temperatures
基于阿伦尼乌斯方程,对SiO2在CaO表面的扩散系数与温度的关系曲线进行动力学拟合,最终得到其扩散活化能。计算所得SiO2在CaO表面上的扩散活化能为28.79 kJ/mol。
2.4 温度对MgO在CaSiO3表面结合、扩散特性的影响
CaSiO3与MgO的配比为60∶40时结合能绝对值最大,因此选取该配比下的模型研究不同温度下MgO在CaSiO3(001)表面的结合能,如表6所示。

表6 不同温度下MgO在CaSiO3(001)表面的结合能Tab.6 Binding Energy of MgO on CaSiO3(001)surface at different temperatures
由表6可知,随着温度的升高,MgO在CaSiO3(001)表面的结合能绝对值逐渐增加,但结合能数值相差不大,尤其是从1 000 ℃到1 100 ℃时的结合能变化非常小。同时,对不同温度下MgO在CaSiO3(001)表面的分子扩散进行分析,得到的均方位移如图11所示。由图11可知,当温度从815 ℃上升到1 100 ℃时,MgO在CaSiO3(001)表面的均方位移随着时间的推移逐渐增大。同时,对均方位移进行线性拟合,求得不同温度下MgO在CaSiO3表面的扩散系数(见表7)。
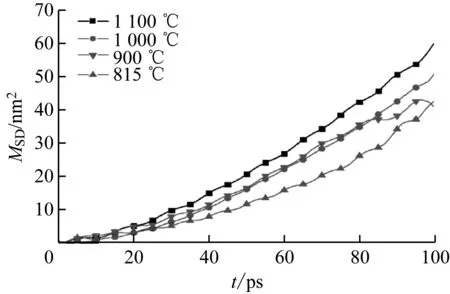
图11 不同温度下MgO在CaSiO3(001)表面的均方位移Fig.11 Mean square displacement of MgO on CaSiO3(001)surface at different temperatures

表7 不同温度下MgO在CaSiO3表面的扩散系数Tab.7 Diffusion coefficient of MgO on CaSiO3 surface at different temperatures
由表7可知,MgO在CaSiO3表面的扩散系数随温度的升高而增大,说明温度越高,MgO分子越易扩散到CaSiO3中。同理可计算得到MgO在CaSiO3表面上的扩散活化能为17.91 kJ/mol。
3 结 论
(1)在CaO与SiO2反应形成CaSiO3的过程中,CaO物质的量占比越高,CaO与SiO2的结合能绝对值越大,越易生成CaSiO3。高钙准东煤灰中的CaO含量极高,导致CaO极易与SiO2反应形成大量的CaSiO3。
(2)CaSiO3与MgO的结合能随着MgO物质的量占比的增加呈先增加后下降的趋势,当CaSiO3与MgO的配比为3∶2时,二者的结合能力最强,越易发生反应形成钙镁黄长石。
(3)温度对CaO在SiO2表面以及MgO在CaSiO3表面结合特性的影响并不明显,而对其扩散具有重要影响。随着温度的升高,CaO在SiO2表面的扩散系数以及MgO在CaSiO3表面的扩散系数均逐渐增大;且MgO在CaSiO3表面的扩散系数明显较大,扩散活化能较小,表明在CaSiO3形成后,MgO将很快扩散到CaSiO3表面,形成钙镁黄长石。
