团簇Co2Mo2P3电子性质的研究
2022-03-26吴庭慧方志刚王智瑶朱依文曾鑫渔
吴庭慧,方志刚,王智瑶,许 友,朱依文,曾鑫渔
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
近年来,科学研究员们致力于新型催化剂[1]的研究,而Pt[2]基是当前催化效率最好的催化剂,但因其价格太高不能被工业生产利用,其他贵金属催化剂[3]也因其在地球上含量少、价格贵等原因都不能工业化生产.Mai[4]等发现Co、Mo和P组成的催化剂具有与贵金属Pt相似的HER[5-7](析氢反应)活性,并且 Co-Mo-P系列合金具有良好的电催化性[8]、稳定性[9]、耐腐蚀性[10]等性质.电化学水分解法[11]通过使用可再生能源在氢燃料生产中引起了相当大的关注.Yang[12]等发现,Co-Mo-P在加氢脱氧反应中表现也很好,以苯酚为模型化合物,以MgO负载的硫化CoMo(含磷和不含磷)作催化剂促进剂,研究了生物粗品的加氢脱氧,发现 CoMoP/MgO在苯酚的加氢脱氧中显示出优异的活性.故文中基于Co-Mo-P系列合金和参考文献[13],以团簇Co2Mo2P3为模型探究团簇的微观电子性质.
1 模型和计算方法
根据拓扑学原理对团簇Co2Mo2P3进行空间立体结构设计,利用密度泛函理论(DFT)[14],在B3LYP/Lanl2dz水平下,对设计的所有初始构型分别于二重态、四重态进行全参数优化计算,逐个排除虚频和相同构型,最终得到8种稳定构型,其中包括4种二重态和4种四重态.利用Multiwfn程序提取各原子的电荷量,并利用Gaussian09程序提取各构型的电子自旋密度与各原子轨道的布居数等相关数据.在B3LYP泛函的条件下,采用Lanl2dz基组对Co的最外层3d74s2价电子、Mo的最外层4d55s1价电子及P的最外层3s23p3价电子进行描述.对Co和Mo金属原子采用Hay[15]等的含相对论校正的有效核电势价电子从头算基组,即采用18-eECP的双ξ基组(3s,3p,3d/2s,2p,2d),P加极化函数为ξPd=0.55[16],所有计算均在计算机M6490上完成.
2 分析与讨论
2.1 团簇Co2Mo2P3构型分析
根据拓扑学原理对团簇Co2Mo2P3进行设计,经过一系列对团簇Co2Mo2P3的优化和计算,最终共得到8种稳定构型如图1所示,分别是三棱双锥戴帽(1(2)、2(4))、六棱锥(1(4)、3(2))、五棱锥(2(2)、3(4)、4(2)、4(4)).其右上方括号内数字代表多重度,设能量最低的构型1(2)的能量为 0 KJ/mol,各重态构型分别按照能量由低到高顺序排列.构型的能量大小排序如下:1(2)<1(4)<2(2)<3(2)<2(4)<3(4)<4(4)<4(2).
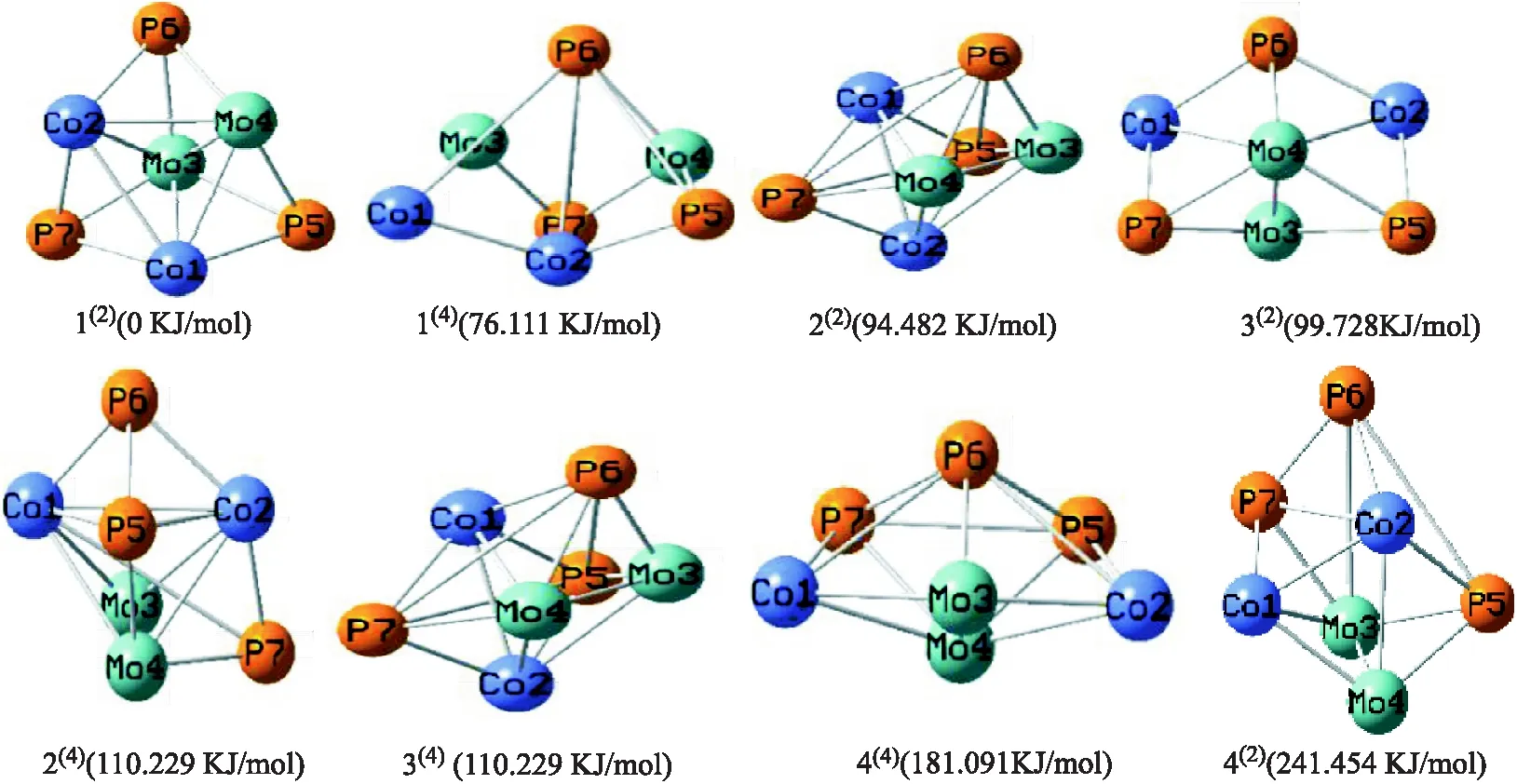
图1 团簇Co2Mo2P3优化构型及能量图
由图1知,团簇Co2Mo2P3的稳定形态有2种,分别是三棱双锥戴帽(1(2)、2(4))、六棱锥(1(4)、3(2))、五棱双锥(2(2)、3(4)、4(2)、4(4)).由上述8种稳定构型的能量大小关系可知:构型1(2)热力学稳定性最好,构型4(2)热力学稳定性最差.
2.2 团簇Co2Mo2P3的电子性质
2.2.1 团簇Co2Mo2P3各原子电荷量
各原子电荷量可以有效地反应出各构型内部原子的电子流向问题,团簇Co2Mo2P3的各稳定构型的原子电荷量见表1.ΣCo表示团簇Co2Mo2P3中2个Co原子电荷量总和,ΣMo为2个Mo原子电荷量总和,ΣP为3个P原子电荷量总和.原子电荷量为正值表示电子从该原子流出,原子电荷量为负值代表有电子流入到该原子.从表1可知,各构型中的P原子电荷量均为正值,这说明P原子是团簇Co2Mo2P3的电子供体.Co原子电荷量除了构型1(4)外均为负值,并且构型1(4)中的Co原子电荷量较小影响不是很大,所以认为Co原子是团簇Co2Mo2P3的电子受体.Mo原子的情况有些复杂,在二重态下,构型1(2)和构型4(2)中Mo原子电荷量为正值,构型2(2)和构型3(2)中Mo原子电荷量为负值,所以电子流向要分情况讨论.而在四重态下,除构型2(4)外均为负值,表示除构型2(4)外Mo原子为电子受体.

表1 团簇Co2Mo2P3各原子电荷量
为了进一步探究团簇Co2Mo2P3的各构型电子流动情况,作出图2.以构型为横坐标,总电荷量为纵坐标,作出各构型Co、Mo、P原子的电荷总量变化趋势图.电子流动性强弱的重要依据是各构型原子间的电荷量改变量,从图2可以明显看出,各个构型中的P原子均为正值,并且P、Co原子的电子流动的幅度很大,故P、Co原子为团簇Co2Mo2P3内部电子流动的主要贡献者.结合原子内电子流动方向和各原子电荷量变化趋势,可以得出结论团簇Co2Mo2P3中各构型的电子流动性强弱关系如下:2(2)>3(4)>4(4)>4(2)>3(2)>2(4)>1(2)>1(4).

图2 团簇Co2Mo2P3原子电荷总量
2.2.2 团簇Co2Mo2P3各原子轨道布居数变化量
团簇Co2Mo2P3的各原子轨道的布居数变化量见表2.电荷在各个原子轨道上都有1个分布(又称布居),布居数表示原子在团簇中各轨道上的电子排布较原子本身电子排布的变化量.布居数变化量的数值为正时,代表该轨道有电子流入;当变化量的数值为负值时,代表该轨道有电子流出.从表2中可以得出:团簇Co2Mo2P3的Co原子的3d与4p轨道、Mo原子的5p轨道、还有除构型2(2)、3(4)、4(2)的P原子3p轨道为负值外,其余构型P原子的3p、3d轨道上的布居数变化量数值均为正值,代表上述轨道有电子流入;Co原子的4s轨道、Mo原子的5s轨道、P原子的3s轨道上的布居数变化量数值均为负值,代表上述轨道有电子流出.从整体上看,团簇内部电子主要是由各原子的s轨道流向p、d轨道.团簇Co2Mo2P3中P原子的s、p、d轨道布居数总变化量为负值,这代表在团簇中P原子为电子供体,这与上述2.2.1所得结论一致.Co原子是在构型1(4)、3(2)中的布居数总变化量为负值,代表有电子流出,提供电子;除1(4)、3(2)构型中外其他构型中Co原子的布居数总变化量为正值,电子从P→Co、Mo.Mo原子是在构型1(2)、2(4)、4(2)中布居数总变化量为负值,代表它们提供电子,有电子流出;在其余构型中Mo原子的布居数总变化量为正值,电子从P→Co、Mo.此外,从表2中明显发现在团簇Co2Mo2P3中的Co原子的4s轨道、Mo原子的5p轨道还有P原子的3p轨道的布居数变化量较大,为团簇Co2Mo2P3内部电子流动的主要贡献者.
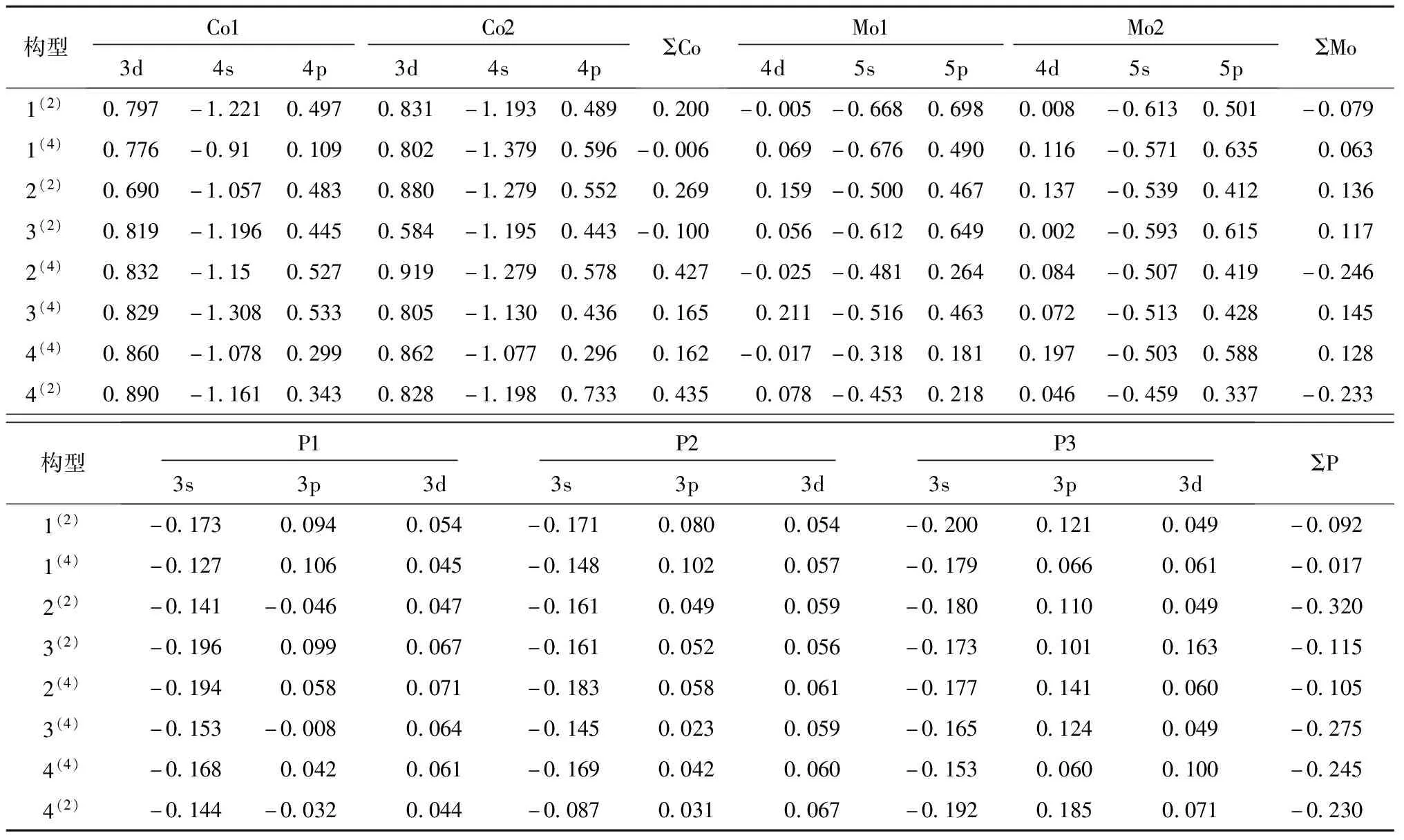
表2 团簇Co2Mo2P3各构型布居数变化量
2.3 团簇Co2Mo2P3的电子自旋密度
2.3.1 团簇Co2Mo2P3各原子的电子自旋密度
各原子的电子自旋密度是影响团簇Co2Mo2P3稳定性的重要因素之一,为了进一步分析将团簇Co2Mo2P3各原子自旋密度数据整理在表3.团簇Co2Mo2P3中各原子自旋密度的正值和负值分别代表α电子出现的净概率密度和β电子出现的净概率密度.从表3中可以看出在构型2(2)-4(2)中Co1、Co2原子的电子分布为自旋向上的α电子.在构型1(2)-3(2)中皆有一个Co原子的电子分布为α电子,一个Co原子的电子分布为β电子.在构型1(2)、3(2)、2(4)、3(4)、4(4)中皆有1个Mo原子的电子分布为α电子,1个Mo原子的电子分布为β电子;在构型1(4)、2(2)中Mo原子的电子分布皆为自旋向上的α电子.P原子在构型1(2)、1(4)、3(4)、4(4)、4(2)中皆有1个P原子的电子分布为α电子,2个P原子的电子分布为β电子.从表中发现,稳定构型1(2)、1(4)中的Co原子1个自旋密度较大、1个自旋密度较小且相差很大,说明2个构型的Co原子可能存在拮抗作用,抑制了2个构型的团簇稳定性.能量一样的构型2(4)和3(4),Co原子的电子分布皆为自旋向上的α电子,且Co原子、P原子的自旋密度数值相差不多,所以构型2(4)、3(4)的稳定程度也是相近的.

表3 团簇Co2Mo2P3各原子的电子自旋密度
2.3.2 团簇Co2Mo2P3各原子间电子自旋密度
各原子间的电子自旋密度是反应团簇Co2Mo2P3稳定性的因素之一,原子间的电子自旋密度是判断各构型原子间成键强度的重要依据,原子间电子自旋密度的绝对值可以反映原子间的成键强度.团簇Co2Mo2P3各原子间的电子自旋密度具体数据如表4,团簇各原子间的电子自旋密度数值的正负分别代表两原子成键时α和β的电子过剩情况.数值为正值时表示两原子成键时有α电子过剩,数值为负值时表示2原子成键时有β电子过剩.从表4中可以看出,最稳定构型1(2)的Co1-P1、Co1-P2、Co1-P3、Co2-Mo1、Co2-Mo2、Co2-P1、Mo1-P1、Mo1-P2、Mo1-P3、Mo2-P1、Mo2-P2、P2-P3成键时为β电子过剩,Co1-Co2、Co1-Mo1、Co1-Mo2、Co2-P2、Co2-P3、Mo1-Mo2、Mo2-P3、P1-P3、P1-P2成键时为α电子过剩.且构型1(2)的各原子间的电子自旋密度的绝对值是8个构型中较大的2个构型,成键强度越大,成键越均匀,所以构型1(2)的稳定性最好,团簇体系的能量最低.最不稳定构型4(2)成键时β电子过剩的键为Co1-P1、Co1-P2、Co1-P3、Co2-P1、Co2-P2、Co2-P3、Mo1-Mo2、Mo1-P1、Mo1-P2、Mo1-P3、Mo2-P1、P1-P3、P2-P3,α电子过剩的键为Co1-Co2、Co1-Mo1、Co1-Mo2、Co2-Mo1、Co2-Mo2、Mo2-P2、Mo2-P3、P1-P2.
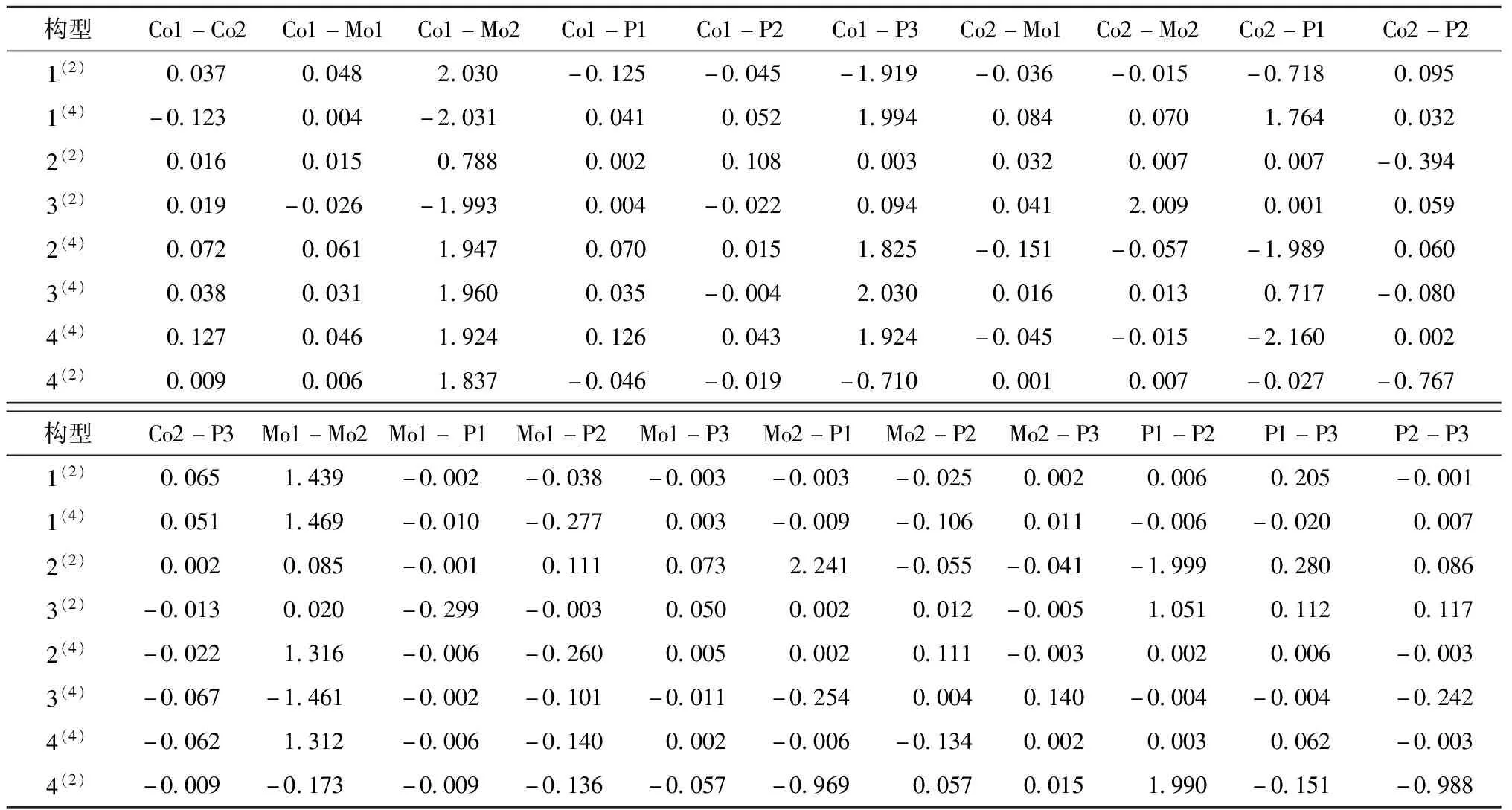
表4 团簇Co2Mo2P3各原子间电子自旋密度
2.3.3 团簇Co2Mo2P3的电子自旋密度图分析
电子自旋密度图是反映团簇稳定性的参考标准,为进一步研究团簇Co2Mo2P3的稳定性,作出团簇Co2Mo2P3的电子自旋密度图3.图中浅色表示α电子,深色表示β电子.从图3中看出构型1(2)和构型1(4)的内部α电子和β电子的重叠部分较多且分布更均匀,使体系能量降低,稳定性更高.每个构型的α电子和β电子都有一定的重叠部分,但重叠部分每个构型都存在一定的差异,所以各个构型的能量也不尽相同,稳定性也不同.构型3(2)和构型3(4)是不同重态下的同一构型,2个构型的电子自旋密度图中的α电子和β电子分布较为接近,重叠区域也相似,因此2个构型之间的能量相差不大,稳定性也接近.从8个稳定构型电子自旋密度图整体来看:每个构型的α电子和β电子皆有一定的重叠部分,但是重叠部分的大小和均匀性却有一定的差异;每个构型的α电子和β电子自旋密度比例也不尽相同.

图3 团簇Co2Mo2P3各构型电子自旋密度图
3 结语
从4个方面分析了团簇Co2Mo2P3的电子流动情况,分别是原子电荷量、布居数、原子间的电子自旋密度、自旋密度图.(1)电荷角度分析得出结论:P原子是团簇Co2Mo2P3的电子供体,是构型内部电子的主要贡献者.(2)布居数角度分析得出结论:团簇内部电子主要是由各原子的s轨道流向p、d轨道,其中团簇Co2Mo2P3中的Co原子的4s轨道、Mo原子的5p轨道还有P原子的3p轨道的布居数变化量较大,所以它们为团簇内部电子流动的主要贡献者.(3)自旋密度角度分析得出结论:团簇Co2Mo2P3中的的α电子和β电子的重叠情况可以影响团簇的稳定性.综上所述,通过对团簇的电子性质进行分析,得出构型1(2)的稳定性是8个构型中最好的结论.对团簇Co2Mo2P3的电子性质分析,为团簇的稳定性研究提供了更多理论支撑.
