叶酸修饰的聚(N-羟丙基甲基丙烯酰胺)-巯基嘌呤纳米粒子的制备和细胞毒性研究
2022-03-20狄钰馨张加赵敏智龙海涛许卫兵
狄钰馨,张加,赵敏智,龙海涛,许卫兵
(甘肃农业大学理学院,甘肃 兰州 730070)
恶性肿瘤是危害人类身体健康的重大疾病,2020 年我国癌症死亡人数达300 万。早期的诊断和有效的治疗对于恶性肿瘤病人来说尤为重要[1]。近年来,肿瘤微环境响应的聚合物抗癌药物输送系统引起广泛关注,如肿瘤部位较低的pH 值和较高的谷胱甘肽含量等都被用于设计相应的响应性抗癌药物输送系统[2-3]。大分子载体由于其在肿瘤组织中的渗透性和滞留作用,被广泛研究成为理想的靶向给药系统[4]。大多数载药系统利用聚乙二醇(PEG)、聚乳酸-羟基乙酸共聚物(PLGA)、聚己内酯(PCL)和聚氨基酸等4 种载体,以被动靶向的方式选择性地将抗癌药物递送至肿瘤[5-6]。聚N-(2-羟丙基)甲基丙烯酰胺(HPMA)共聚物作为一种水溶性、无毒、无免疫和生物相容性好的亲水组分,含有羟基的聚(HPMA)可以作为载体,通过酯键连接各种药物,进而构建潜在的肿瘤微环境响应性的药物传递系统已得到广泛研究[7-8]。叶酸是人体所必需的一种B 族维生素,与正常细胞相比,肿瘤细胞由于不受控制的分裂而需要摄取更多叶酸,因此多种肿瘤细胞表面都过度表达着叶酸受体,叶酸受体能够与叶酸分子发生特异性识别[9],两者之间有很强的亲和力,通过这种特殊作用,可将与叶酸结合的药物分子或载药体系靶向性送入肿瘤细胞中[10-11],叶酸修饰的纳米载药系统可以靶向性将药物输送到特定细胞膜,并使得纳米粒子被内吞到肿瘤细胞中[12],因此叶酸被作为癌细胞靶向配体而受到广泛研究[13]。
6-巯基嘌呤(6-MP)是一种硫鸟嘌呤衍生物,广泛用于临床治疗急性淋巴细胞白血病。其生物活性机制可能涉及多种细胞代谢调节。一般认为,6-MP的毒性活性主要来自于被纳入核酸的代谢核苷酸。此外,它还可以作为嘌呤核苷酸从头合成的伪反馈抑制剂,通过与正常嘌呤竞争形成抑制性类似酶复合物,从而破坏必要中间产物的合成和嘌呤核苷酸的相互转化[14-15]。3-(7H-嘌呤-6-硫基)丙烯酸(PTA)是以6-MP为母体而设计的低毒性小分子前药,含有α,β 不饱和硫代羧酸酯结构,其中顺式的PTA能在谷胱甘肽的作用下,经历加成-消除反应转变形成6-MP[16],从而起到抑制癌细胞生成的作用。癌细胞由于不断的分裂活动,细胞中谷胱甘肽的含量是正常细胞中的数倍,因此PTA前药在正常细胞中不会释放出6MP,对正常细胞毒性较低。利用PTA上的羧基,将该前药共价链接到聚(HPMA)上制备成的纳米高分子抗癌药物[17-18],提高药物的水溶性和利用度,在高分子药物进入癌细胞后,通过与谷胱甘肽(GSH)发生加成消除反应而释放6-MP,从而抑制肿瘤的分裂,达到抗癌的目的。
基于以上分析,本实验首先利用活性聚合的方式制备了特定分子量的聚(HPMA),然后利用叶酸(FA)和PTA分子中的羧基与聚(HPMA)分子链上的羟基之间的反应,将FA 和PTA 同时连接于聚(HPMA)上,构建了具有主动靶向功能的聚合物抗癌药物聚(HPMA)-FA-PTA,未连接FA 的聚(HPMA)-PTA 作为对照聚合物,利用核磁共振氢谱、傅里叶变换红外光谱仪和透射电镜等对聚合物的结构进行了表征检测,随后对其细胞毒性和体内靶向性进行了进一步研究。
1 材料与方法
1.1 材料与试剂
RPMI1640 培养基、3-(4,5-二甲基噻唑-2)-2、5-二苯基四氮唑溴盐(MTT)、胎牛血清(FBS)购自北京索莱宝科技有限公司;二甲基亚砜(DMSO)购自天津恒兴化学试剂制造有限公司,罗丹明B、DTT(C4H10O2S2FW)、还原型谷胱甘肽(C10H17N3O6S)购自上海阿拉丁生物化学技术有限公司。聚(HPMA)为本实验室自制。
1.2 仪器
电感耦合等离子体质谱(ICP-MS)美国Varian公司;JSM-5510LV 型扫描电子显微镜(SEM),JEOLJEM-2100 高分辨率透射电子显微镜(TEM,HRTEM,200kV);动态光散射(DLS)和Zeta粒度分析仪(SALD,2300),日本岛津公司;傅里叶变换红外(FT-IR)吸收光谱仪Galaxy7020A,马特森仪器公司;赛默飞世尔科技公司;紫外吸收光谱仪;Netzsch STA 409 PC analyzer 热重分析仪;细胞培养箱(NU-4750E型,CO2孵箱),美国Nuarie公司;酶标仪(RT-6100),深圳雷杜生命科学股份有限公司。
1.3 3-(7H-嘌呤-6-硫基)丙烯酸(PTA)的制备
将6-MP(0.510 g,3.0 mmol)溶解于25 mL甲醇中,加入甲醇钠(0.594 g,11.0 mmol),搅拌至以上两种化合物完全溶解后,加入丙炔酸(0.185 mL,3.0 mmol),将该溶液置于65 ℃油浴中回流过夜,向溶液中加入4 mL 蒸馏水进行淬灭,然后滴加1.0 mol/L 的盐酸形成沉淀,抽滤得到粗产品,将粗产品溶解于1.0 mol/L 的氢氧化钠溶液,再加入1.0 mol/L的盐酸形成沉淀,抽滤得到纯产物PTA。
1.4 聚(HPMA)-FA-PTA的制备
将0.2 g 聚(HPMA)、0.03 g PTA 和0.02 g FA溶解于2 mL DMSO 中,待其完全溶解后,将该溶液置于冰水浴中冷却到0 ℃,再加入二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP),在氮气保护下反应48 h后,用丙酮和乙醚的混合液沉淀,过滤沉淀并将其溶解于10 mL 的甲醇中,最后用截留分子量为3 000 的超滤浓缩离心管离心得高分子化合物聚(HPMA)-FA-PTA。聚(HPMA)-PTA 的制备方法与聚(HPMA)-FA-PTA 的制备方法类似,将制备过程中的FA去掉即可制得。
1.5 细胞培养
人肝癌细胞HepG2 用RPMI1640 培养基培养,每个培养基中添加10% 胎牛血清(FBS)和100 U/mL 青霉素,链霉素0.1 mg/mL,将细胞保存在5% CO2,37 ℃的培养箱中培养。
1.6 细胞毒性试验
采用MTT法测定细胞存活率,以评价所制备的聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 的体外细胞毒性。将HepG2 细胞以5×103/孔的密度接入96 孔板中,于37 ℃和5% CO2条件下培养过夜使细胞贴壁生长。然后加入不同浓度的聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 溶液,其中含6-MP分别为2.5,5,10,40,80 mg/L,培养24 h 和48 h 后,每孔加入20 μL MTT(5 mg/mL)在培养箱中孵化,4 h 后弃掉96 孔板的培养基并每孔加入150 μL DMSO并震荡10 min,在490 nm的酶标仪下记录其吸光度(OD),以生理盐水OD值为对照,计算细胞存活率。
1.7 细胞成像测定
将HepG2细胞加到六孔板(1×105个/孔)上,每孔2 mL,然后将细胞培养于5% CO2,37 ℃的培养箱中培养24 h 后,分别加入聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 溶液,确保药物的终浓度为25 mg/L 和100 mg/L。加药后将细胞保存在5%CO2,37 ℃的培养箱中培养24 h后弃掉培养基,每孔用1 mL PBS 清洗2 次,弃掉PBS,每孔加500 μL 固定液固定15 min后弃掉固定液,每孔加1 mL PBS清洗2 次。在离心管中避光配染色液(490 μL PBS,5 μL Hochest33342,5 μL PI 混 合),每 孔 加 入5 μg/mL 染色液轻摇15 min 后弃掉染色液,每孔加入1 mL PBS 清洗,每次洗7~8 min,重复3 次,每孔加1 mL PBS 后在荧光显微镜下镜检。每个浓度设置3个复孔,PBS处理组作为对照。
1.8 溶酶体逃逸测定
首先用罗丹明B 标记浓度为20、50 mg/L 的聚(HPMA)-PTA,聚(HPMA)-FA-PTA聚合物孵育细胞24 h,然后用溶酶体特异性荧光染料浸泡细胞2 h,在荧光显微镜下观察聚合物在细胞中的分布情况。
1.9 细胞凋亡测定
采用annexin V-FITC 凋亡检测试剂盒定量凋亡和坏死细胞,评价细胞死亡机制。将HEPG2细胞加到六孔板(1×105/孔)中,每孔2 mL,共接4 个孔后将细胞保存在5% CO2,37 ℃的培养箱中培养24 h,抽出1 mL 培养基后弃掉,再每孔加入1 mL 新的培养基。分别将25、100 mg/L 的聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 加入到4个孔中,将细胞保存在5% CO2,37 ℃的培养箱中培养48 h,现场处理。收集细胞上清液,用1 mL PBS清洗后收集上清液,用300 μL胰酶消化六孔板壁上的残留细胞,晃动并拍打后收集上清液。再用1 mL PBS 清洗两次后收集上清液。在离心机1 500 r/min 离心5 min后弃掉上清液。从侧壁加500 μL PBS 再次离心后弃掉上清液,重复2 次。在每个离心管中加300 μL结合液,避光每管加5 μL Annexin-V后等15 min,避光加入10 μL PI 后等待5 min。把离心管中的液体全部加入六孔板后在流式细胞仪上镜检。
2 结果与分析
2.1 制备与表征
首先利用可控活性聚合制备了聚(HPMA),详细的制备过程参考本实验室已发表的相关文献[19],聚(HPMA)上包含有大量可反应性羟基。本试验利用FA和PTA中的羧基与羟基之间能够形成酯键的原理,利用DCC 和DMAP 分别作为脱水剂和催化剂,将靶向配体FA 和药物PTA 同时连接于聚(HPMA)上,具体的制备过程如图1所示。

图1 聚(HPMA)-PTA、聚(HPMA)-FA-PTA的制备过程Figure 1 Preparation process of poly (HPMA)-PTA and poly (HPMA)-FA-PTA
所制备聚合物的1H NMR 谱如图2 所示。对于聚(HPMA)-PTA,化学位移位于δ=3.67处的质子信号归属于聚(HPMA)上与羟基相连的次甲基(-CONH-CH2-CH(OH)-CH3),而与酰胺直接相连的亚甲基(-CONH-CH2-CH(OH)-CH3)的质子信号出现在δ=2.91,聚合物主链上的甲基、亚甲基和与次甲基相连的甲基(-CH(OH)-CH3)则出现在化学位移δ位于0.5~2.0的范围内,PTA结构的特征质子的信号出现在δ 为8.4~8.8 和6.4~6.6。聚(HPMA)-FA-PTA 中除了出现PTA 的特征化学位移外,在δ为6.4和7.6处分别出现了FA中苯环的特征质子信号,更详细的归属标记于图2中。由此可以看出,PTA和FA均成功连接到聚(HPMA)上,成功制备了聚(HPMA)-PTA 和聚(HPMA)-FAPTA 两种载药聚合物。FA 和PTA 在聚(HPMA)-FA-PTA 中的含量分别是用叶酸(8.83)中杂环氢的积分面积,PTA 中不饱和氢(5.68)的积分面积和HPMA(3.68)中H3CH(OH)CH2NH 氢的积分面积来计算的。聚(HPMA)-FA-PTA 分别含有5.8%和9.4%的FA和PTA。

图2 聚(HPMA)-FA-PTA(a)、聚(HPMA)-PTA(b)的核磁共振氢谱Figure 2 1H NMR spectra of poly (HPMA)-PTA and poly (HPMA)-FA-PTA
利用红外表征了所制备样品的官能团的变化(图3),对于纯的PTA,位于3 430 cm-1处的峰归属于结构中羟基的不对称伸缩振动,位于3 010~3 100 cm-1处的峰归属于嘌呤环上亚甲基的不对称伸缩振动,2 980 和2 810 cm-1处的峰对应于烷基上的亚甲基和次甲基的不对称伸缩振动,其中羰基的振动吸收峰则出现在1 710 cm-1,嘌呤环的伸缩振动则出现在1 510 cm-1处,位于1 050~1 350 cm-1范围内的大量吸收峰则归属于C-O,C-C,C-N和C-S键的不对称伸缩振动。对于未经过任何修饰的聚(HPMA),其结构中羟基的伸缩振动峰出现在3 300 cm-1处,1 630 和1 510 cm-1处对应羰基的伸缩振动和NH 基团的弯曲振动,聚(HPMA)-PTA、聚(HPMA)-FA-PTA 的红外图谱与聚(HPMA)的图谱相似。这可能是由于PTA和FA两种基团的含量较少所导致的。

图3 聚(HPMA)-PTA、聚(HPMA)-FA-PTA的FT-IR光谱Figure 3 FT-IR spectra of poly (HPMA)-PTA and poly (HPMA)-FA-PTA
聚(HPMA)、聚(HPMA)-PTA、聚(HPMA)-FA-PTA的热重曲线如图4所示。3种聚合物均在100 ℃以下出现大约6%的失重,主要归属于吸附在聚合物表面的水分蒸发。聚(HPMA)在200~600 ℃之间出现连续的热失重过程,其DTG曲线上分别在350 ℃和450 ℃出现明显的放热峰,这是由于聚合物中羟基的氧化、脱水、缩合以及骨架的燃烧造成的。由于热重测试在空气气氛中进行,因此当温度高于600 ℃时聚合物最后剩余质量基本为0%,表明聚合物完全燃烧。聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 的热重曲线和聚(HPMA)的曲线基本相似,而聚(HPMA)-PTA 的DTG 曲线中位于350 ℃的放热峰虽然与聚(HPMA)的出峰位置相同,但是该峰的面积明显增大,第2个放热峰的位置由450 ℃降至410 ℃,同时位于580 ℃的第3个放热峰也降低至570 ℃,可以发现这两个放热峰的面积均有所减小,以上变化表明PTA 成功的引入到聚(HPMA)中。而在聚(HPMA)-PTA 中引入FA 后,位于350 ℃的放热峰大规模的降至接近200 ℃,且峰面积进一步增大,位于410 ℃的放热峰则没有出现明显的变化,位于580 ℃的放热峰降低至520 ℃,且峰面积明显增加,以上变化表明FA 被成功引入到聚合物中。
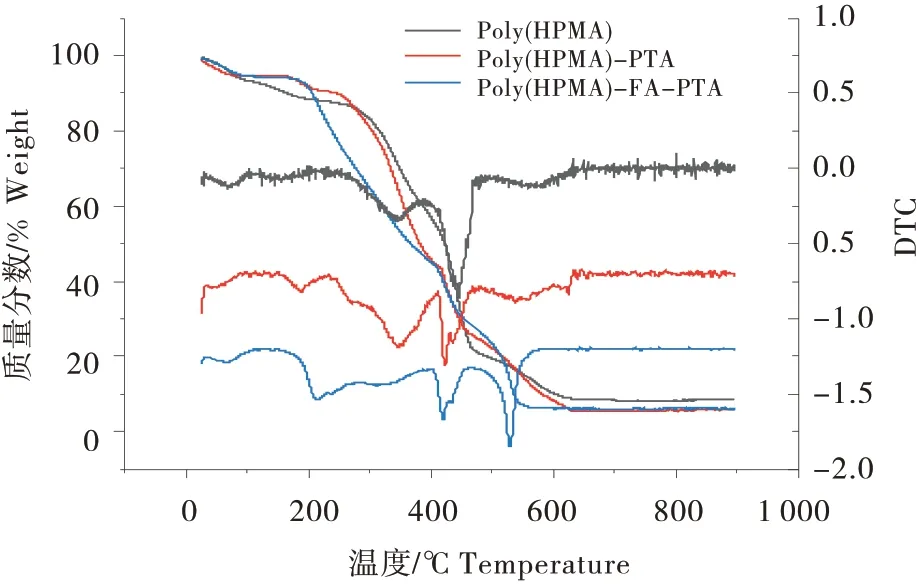
图4 聚(HPMA)-PTA、聚(HPMA)-FA-PTA的热重曲线Figure 4 The TGA of poly (HPMA)-PTA and poly(HPMA)-FA-PTA
由于聚(HPMA)-PTA 和聚(HPMA)-FAPTA两种药物粒子都具有双亲性,因此可以在水溶液中有自组装的性能,两种载药粒子在溶液中的稳定性如图5所示。聚(HPMA)-PTA 5 d的平均粒径在183 nm到178 nm之间波动。而聚(HPMA)-FAPTA 5 d 的平均粒径在90 nm 到71 nm 范围之间波动,变化幅度不大,两种载药粒子都表现出良好的分散性和稳定性。聚(HPMA)-PTA 连续测试5 d 的电位值在-9.39~-7.8 mV 之间波动,而聚(HPMA)-FA-PTA 在相同时间内的电位值在-20.16~-18.09 mV之间波动,这可能是由于叶酸的引入所导致的,可以看出两种粒子的电位值都为负值,这将有利于载药颗粒在生物体内的循环和发挥作用,较小的电位波动同样表明材料具有良好的稳定性。

图5 聚(HPMA)-PTA、聚(HPMA)-FA-PTA的粒径和电位随时间的变化图Figure 5 Plots of particle size and potential with time for poly(HPMA)-PTA,poly(HPMA)-FA-PTA
用透射电镜对聚(HPMA)-PTA、聚(HPMA)-FA-PTA两种样品的微观结构进行观察(图6),可以清楚地观察到两种聚合物都能够组装为球形的纳米粒子,且球形结构较为规则,这是由于两种聚合物的双亲性所引起的,在水溶液中容易自组装成纳米结构。图中还可以发现少量的直径较大的粒子团聚体,这是由于纳米粒子的表面能较大,容易团聚所引起的,样品制备过程中主要通过超声波去除溶液中的微小气泡,由于去除不够完全,也是导致纳米颗粒团聚的原因。其中聚(HPMA)-PTA形成的粒子的直径在100~150 nm 左右,而聚(HPMA)-FA-PTA的直径约为70~90 nm,这是由于聚(HPMA)-FAPTA中包含更多的疏水性成分,导致自组装形成的纳米粒子直径减小,这与两种纳米粒子的粒度数值相一致。
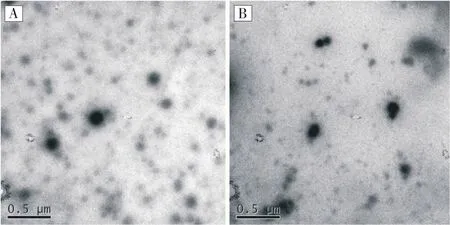
图6 聚(HPMA)-PTA的透射电镜和聚(HPMA)-FAPTA透射电镜Figure 6 Transmission electron microscopy (HPMA)-PTA and poly (HPMA)-FA-PTA
2.2 细胞毒性检测
利用MTT方法,选以HepG2细胞作为模型,研究了复合纳米粒子的体外毒性,聚(HPMA)-PTA和聚(HPMA)-FA-PTA 的浓度都设置为(25、50、100、400、800 mg/L),孵育24 h和48 h后的细胞存活率变化见图7。由图7 可以看出两种复合纳米粒子对细胞表现出显著的抑制作用,两种粒子在低浓度时对HepG2细胞的抑制活性基本与对照组相同,在25 mg/L孵育24 h,聚(HPMA)-PTA和聚(HPMA)-FA-PTA 细胞存活率为分别94.26%和77.37%。相同浓度下,孵育48 h细胞存活率有所下降,分别为91.53%和73.75%。随着浓度增大和时间的增加,细胞的存活率则逐渐下降。浓度达到800 mg/L 孵育24 h,聚(HPMA)-PTA 和聚(HPMA)-FA-PTA细胞存活率下降到69.45%和68.72%。当孵育时间延长至48 h,细胞存活率下降到40.16%和43.59%。聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 在24 h的IC50分别为3.633 和3.554,而48 h 的下降到3.162和3.022(表1)。可以看出浓度越大,孵育时间越长,细胞存活率越低。
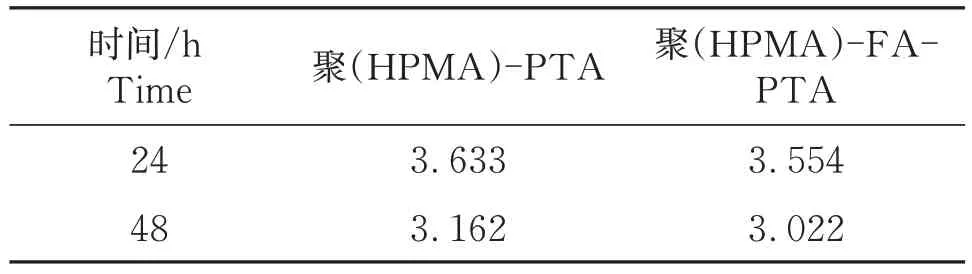
表1 聚(HPMA)-PTA、聚(HPMA)-FA-PTA的IC50值Table 1 IC50 value of poly (HPMA)-PTA and poly(HPMA)-FA-PTA
为了观察药物作用后细胞核结构的改变,首先将HepG2 细胞与聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 孵育24 h,然后用特异性荧光染料Hoechst 33342和碘化丙啶(PI)染色。图8显示了聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 在不同浓度下HepG2细胞的荧光图像。未加药的细胞表现出普通圆形或椭圆形,24 h后观察荧光分布均匀,细胞核大小和轮廓清晰,视野内可见大量细胞,表面细胞生长良好。以25 mg/L 的聚(HPMA)-PTA、聚(HPMA)-FA-PTA处理24 h,少数细胞细胞核不规则。但浓度增加到100 mg/L时,视野内细胞数量明显减少,细胞数量明显减少,细胞核形态越来越不规则,出现核碎裂现象。随着药物浓度的增加,细胞核的体积逐渐增大。综上所述,6-MP通过氧化还原反应与聚HPMA连接,从而显著提高药物利用率。

图8 细胞双染Figure 8 Cell double staining
图9可以清楚地看到聚合物被罗丹明标记后呈现红色荧光,被溶酶体探针标记后呈现绿色荧光。红色和绿色荧光结合时呈现淡黄色,说明结合物可以逃脱溶酶体捕获,表现出良好的溶酶体逃逸功能,有助于减少药物在溶酶体位点的破坏,促进药物向细胞核传递。

图9 溶酶体逃逸Figure 9 Lysosomal escape
通过皮尔森相关系数(R)来定量描述逃逸溶酶体的能力,当聚(HPMA)-PTA 浓度为20 mg/L 时检测到的R值为0.587,而当浓度上升到50 mg/L时检测到的R值为0.642。同理当聚(HPMA)-FAPTA浓度为20 mg/L时检测到的R值为0.520,而当浓度上升到50 mg/L 时检测到的R值为0.828。随着浓度的增加,聚合物在细胞内的逃逸能力也随之增加。
通过流式细胞术进一步研究聚(HPMA)-PTA、聚(HPMA)-FA-PTA 介导的HepG2 细胞凋亡,探讨其体外细胞毒性作用的机制。将化合物处理HepG2 细胞48 h 后,用Annexin V 和碘化丙啶(PI)染色,评价细胞活性(Annexin V-/PI-)、早期凋亡(Annexin V+/PI-)、晚期凋亡(Annexin V+/PI+)和坏死(Annexin V-/PI+)细胞的百分比。细胞密度如图10 所示。与对照组相比,聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 结合物均可导致细胞凋亡。然而,与对照相比,加入聚合物后细胞的凋亡率升高。聚(HPMA)-PTA 和聚(HPMA)-FAPTA 的细胞毒性分别达到22.01%和32.05%,进一步说明聚合物偶联物对肿瘤细胞的细胞毒性是通过诱导细胞凋亡介导的。其高效性可能是由于聚(HPMA)-PTA 和聚(HPMA)-FA-PTA 对6-MP的释放具有较强的控制能力[20-21]。

图10 对照组(A)和25 mg L-1聚(HPMA)-PTA(B)、聚(HPMA)-FA-PTA(C)孵育后的细胞的流式细胞术分析Figure 10 Flow cytometric analysis of cells incubated with CK (A) and 25mg L-1 poly (HPMA)-PTA (B)and poly (HPMA)-FA-PTA (C)
3 讨论
6-MP 由于其水溶性差、血浆半衰期短、生物利用度不稳定、治疗后出现严重的骨髓毒性和肝毒性等副作用,在临床应用上有一定的限制[22]。因此将小分子6-MP连接到生物相容性好的载体上制备成肿瘤微环境响应的高分子抗癌药物,同时引入具有能与癌细胞特异性识别的靶向配体,将药物精准递送到癌细胞是目前研究的重要领域[23-24]。淀粉、壳聚糖、透明质酸、碳量子点等材料常用来作为载体,聚(HPMA)是一种高分子药物载体,具有良好的生物相容性、水溶性、无免疫原性等优点,不仅能够降低药物的毒副作用,减少抗药性,提高药物体内的稳定性,还可以增加药物在肿瘤部位的累积,使药效得到更好的发挥[25-26]。在癌细胞表面过度表达的叶酸受体,由于和叶酸之间强的特异性识别作用,使得叶酸分子成为出色的靶向配体,被广泛用于靶向药物的递送中。此外癌细胞中谷胱甘肽的浓度是正常细胞的4倍,谷胱甘肽是一种含硫醇的三肽,在生物环境中具有强还原能力[27],因此,常被用来设计氧化还原响应性的药物释放。
本文在课题组前期工作的基础上,利用简单的催化活化反应,成功构建了叶酸靶向的对谷胱甘肽刺激响应型的聚(HPMA)-6MP 高分子载药纳米粒子。核磁共振氢谱测试结果表明叶酸和6-MP的前药PTA都成功地链接到聚(HPMA)的羟基上,并利用特征氢原子积分面积的比例计算出叶酸和PTA的含量约在6%和10%。红外和热重分析进一步证明了其结构的正确性。由于该载药系统的聚(HPMA)为亲水性的,而FA和PTA都为疏水性的,因此其具有典型的双亲性结构,能够在溶液中自组装为纳米粒子,样品的透射电镜测试证明了上述结果,从透射电镜的照片中可以明显看到所制备的聚合物在溶液中确实自组装为球形的纳米粒子,且粒子的团聚程度较轻,两种纳米粒子由于是否包含叶酸,球形粒子的尺寸出现明显的差异。连续多天的测试证明了组装形成的纳米粒子的稳定性良好,粒度、粒度分布和电位均随着时间的变化而未出现较大波动。
体外细胞毒性试验结果显示,叶酸的引入较为明显的增强了载药纳米粒子对HepG2的抑制作用,荧光染色可以看出,与对照组相比,两种聚合物载药纳米粒子均能诱导HepG2的细胞出现细胞核变形和碎裂。随后利用荧光试剂罗丹明对载药聚合物进行了标记并开展了溶酶体逃逸试验,结果显示两种载药粒子在浓度分别为20和50 mg/L 时,皮尔森相关系数均大于0.5,表现出较好的溶酶体逃逸性能[28]。流式细胞仪分析进一步表明,与对照组细胞相比,两种载药粒子能够明显地诱导HepG2细胞凋亡,表现出较好的体外抗癌效果[29-30]。
4 结论
将叶酸和具有谷胱甘肽刺激释放性质的前药PTA连接于聚(HPMA)上,成功制备了具有癌细胞特异性识别性能的小分子药物6-MP 纳米递送体系,其中6-MP的载药量约为10%,在溶液中自组装为尺寸约为100~150 nm 的球形粒子,且稳定性较好。细胞试验结果显示,所制备的载药聚合物能够较好地抑制HepG2的增长,引起细胞核的变形和碎裂,明显增强了对细胞凋亡的诱导,且具有较好的溶酶体逃逸能力,因此,在实现6-MP在肿瘤微环境中的释放、提高其生物利用度以及减少药物的副作用方面具有良好的前景。
