An atypical ubiquitin ligase at the heart of neural development and programmed axon degeneration
2022-03-19SatpalVirdee
Satpal Virdee
Abstract The degeneration of nerve fibres following injury was first described by Augustus Waller over 170 years ago.Initially assumed to be a passive process, it is now evident that axons respond to insult via regulated cellular signaling events resulting in their programmed degeneration.Pro-survival and prodegenerative factors have been identified and their regulatory mechanisms are beginning to emerge.The ubiquitin system has been implicated in the pro-degenerative process and a key component is the ubiquitin E3 ligase MYCBP2 (also known as PHR1).Ubiquitin E3 ligases are tasked with the transfer of the small protein modifier ubiquitin to substrates and consist of hundreds of members.They can be classified as single subunit systems or as multi-subunit complexes.Their catalytic domains can also be assigned to three general architectures.Hints that MYCBP2 might not conform to these established formats came to light and it is now clear from biochemical and structural studies that MYCBP2 is indeed an outlier in terms of its modus operandi.Furthermore, the unconventional way in which MYCBP2 transfers ubiquitin to substrates has been linked to neurodevelopmental and pro-degenerative function.Herein, we will summarize these research developments relating to the unusual features of MYCBP2 and postulate therapeutic strategies that prevent Wallerian degeneration.These have exciting potential for providing relief from pathological neuropathies and neurodegenerative diseases.
Key Words: chemical biology; E3 ligase; MYCBP2; neurodegeneration; progammed axon death;structural biology; ubiqultin; wallerian degeneration
Introduction
First reported over 170 years ago, Wallerian degeneration (WD) is the programmed degeneration of axons following their injury and occurs in the central and peripheral nervous systems.The process is characterized by mitochondrial swelling, cytoskeletal disruption and axon fragmentation(Waller, 1851; Coleman and Hoke, 2020).As peripheral neuropathy is a significant and dose-limiting side-effect of common chemotherapeutics,pharmacological inhibition of WD might not only improve quality of life for cancer survivors, but could increase the efficacy of existing drugs in the clinic (Geisler, 2020).Furthermore, axon degeneration occurs in the early stages of amyotrophic lateral sclerosis, Parkinson’s and other age-related neurodegenerative disorders, hence WD inhibitors may confer benefits in the context of neurodegenerative diseases (Dadon-Nachum et al., 2011; Adalbert and Coleman, 2013; Tagliaferro and Burke, 2016; White et al., 2019).
Despite our understanding of the exact molecular mechanism of WD being incomplete, remarkable progress has been made in this area and pro-survival and pro-degenerative factors have been identified.Hints of the existence of a pro-survival factor came from the striking phenotype observed with a strain of mouse known as slow Wallerian degeneration(WldS), where severed axons survived for weeks after detachment from their cell bodies (Lunn et al., 1989).WldS mice were found to have undergone a chromosomal rearrangement where the gene product responsible was a fusion protein (WldS) containing the (NAD+) biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1).The fusion protein resulted in NMNAT1 being relocalized to axons, suggestive of NAD+levels having a regularity role in axon integrity (Mack et al., 2001; Wang et al., 2005).It was subsequently shown in wild-type mice that levels of the neuronal NMNAT isoform, NMNAT2, are depleted post-injury and knock-down induces Wallerian-like degeneration (Gilley and Coleman, 2010).Furthermore,NMNAT2 overexpression is axon-protective so strategies that activate or stabilize NMNAT2 might have therapeutic value (Coleman and Hoke, 2020).Certain disease links to NMNAT2 levels or activity have also been established as their reduction has been associated with Alzheimer’s disease, paediatric neurological disease and fetal akinesia deformation syndrome (Ali et al., 2016;Huppke et al., 2019; Lukacs et al., 2019).
Amongst the pro-degenerative factors that have been identified in mammals is the protein Sterile Alpha and Toll/interleukin-1 receptor TIR motif containing 1 (SARM1) (Osterloh et al., 2012).Mice constitutively depleted of SARM1 appear healthy and this delays axonal degeneration by several days following axotomy.SARM1 knockout also attenuates Wallerian-like degeneration upon exposure to common chemotherapeutic drugs including vincristine and Bortezomib (Gerdts et al., 2013; Geisler et al., 2019; Geisler,2020).It is now evident that SARM1 mediates its degenerative effect through enzymatic NAD cleavage activity (Essuman et al., 2017).This presents a tangible therapeutic target and inhibitors of SARM1 NAD cleavage activity are in active development (Krauss et al., 2020).PubMed was used to search literature and relevant work in the area of MYCBP2, in consideration of a mini review format, was used as the selection criteria for citations.Dated: October 2021.
Programmed Axon Degeneration Is Positively Regulated by the Ubiquitin System Component MYCBP2
An additional pro-degenerative factor is the protein MYCBP2.MYCBP2 is conserved fromC.elegansthrough to mammals and the various orthologues go by a multitude of names (C.elegans Rpm-1, Drosophila Highwire, zebrafish Esrom, mouse Phr1 and human Pam).Collectively they have been coined PHR proteins (Pam/Highwire/Rpm-1) (Grill et al., 2016).MYCBP2 was first identified as a huge 510 kDa interactor with the proto-oncogene Myc (Guo et al.,1998).Although the physiological significance of this interaction has not been elaborated, genetic screens in Drosophila andC.elegansidentified Highwire and Rpm-1 as important regulators of synaptogenesis (Schaefer et al., 2000;Wan et al., 2000; Zhen et al., 2000).A conserved role in synaptic development was also found in vertebrates as nerve terminal morphology is severely disrupted in zebrafish and mice constitutively lacking Esrom and MYCBP2,respectively (Burgess et al., 2004; D’Souza et al., 2005; Bloom et al., 2007).In mice the phrenic nerve does not fully innervate the diaphragm resulting in perinatal lethality due to respiratory distress.However, null and hypomorphic mutants of Drosophila Highwire were found to strongly inhibit axonal degeneration after axotomy (Xiong et al., 2012).Furthermore, conditional silencing of MYCBP2 in adult mice is tolerated for at least 6 weeks and confers axon-protective effects similar to those observed with loss of SARM1 (Babetto et al., 2013).Protection is observed in response to lesion of sciatic nerve andretinal ganglion neurons, illustrating the protective effects of MYCBP2 loss in both the peripheral and central nervous system.It has also recently been shown that loss of Highwire in Drosophila protects dopaminergic neurons and improves survival after traumatic brain injury; a major cause of human death and disability worldwide (Hill et al., 2020).Thus, despite the importance of MYCBP2 in neurodevelopment, post-developmental inhibition might be tolerated in humans.Particularly when applied to chemotherapy-induced neuropathy and neuron trauma, where acute inhibition would be sufficient.
MYCBP2 is a multi-domain E3 ubiquitin ligase (termed E3 hereon), a class of enzyme that catalyses the covalent conjugation of the small protein ubiquitin to target proteins (Zheng and Shabek, 2017).There are more than 600 ubiquitin E3s, either single polypeptides or multi-subunit complexes, that operate at the end of an enzymatic cascade involving initiating E1 activating enzymes and intermediary E2 conjugating enzymes (Hershko and Ciechanover,1998) (Figure 1).Ubiquitination performs a wide range of cellular functions with regulation of protein substrate stability by proteasomal degradation being most notable (Pohl and Dikic, 2019).The pro-degenerative effects of MYCBP2 have been linked to its ability to destabilize NMNAT2 by neuronal proteasomal degradation (Xiong et al., 2012; Babetto et al., 2013; Desbois et al., 2018).Hence, inhibition of MYCBP2 E3 activity might be a strategy for inhibiting WD and preserving axon integrity.However, whether inhibition of MYCBP2 E3 activity specifically would have an axon-protective effect has remained unclear as neuronal phenotypes and cellular localization have been attributed to MYCBP2 regions outside the C-terminal ubiquitin ligase module(Grill et al., 2007; Abrams et al., 2008; Dorr et al., 2015).

Figure 1 | Cysteine-dependent ubiquitin ligases are key components of the ubiquitinproteasome system.
MYCBP2 Is a Novel RING-Cys-Relay Ubiquitin Ligase
In an unexpected discovery we showed that MYCBP2 is the sole member of a highly unusual class of E3 as at its extreme C-terminus resides an unprecedented 30 kDa enzymatic module termed RING-Cys-relay (RCR) (Figure 2A) (Pao et al., 2018).This was surprising because it was generally accepted that the different classes of catalytic E3 module had already been established.This discovery was enabled by chemical biological tools known as activitybased probes (Mulder et al., 2020).The activity-based probes covalently label E3s that demonstrate the transthiolation activity mediated by E3s that utilize a catalytic cysteine nucleophile (Figure 1) (Pao et al., 2016, 2018).Approximately 45 transthiolation E3s were believed to exist and MYCBP2 is a new addition to this subtype.
A highly unusual characteristic of MYCBP2 is that unlike other classes of transthiolation E3 it contains a second, downstream catalytic cysteine residue(Figure 2B).Once the initial covalent ligase-ubiquitin intermediate has been formed, the Ub molecule is intramolecularly “relayed” over a distance of ~24 Å to a downstream site.It is this downstream site that is tasked with substrate modification (Pao et al., 2018).Both of the catalytic cysteines reside within an unprecedented zinc ion-binding protein domain termed the tandem cysteine domain (Figure 2C).Further irregularities pertain to MYCBP2 because E3s studied previously invariably couple Ub to lysine amino groups via an isopeptide bond.In a striking digression from this dogma, MYCBP2 couples Ub to hydroxyl groups via an ester bond, where a strong preference is observed for threonine over serine residues in model substrates.Not only does this highlight the unappreciated substrate scope of ubiquitination, but these distinct characteristics of the RCR module might facilitate the development of specific small molecule inhibitors of MYCBP2 E3 activity.
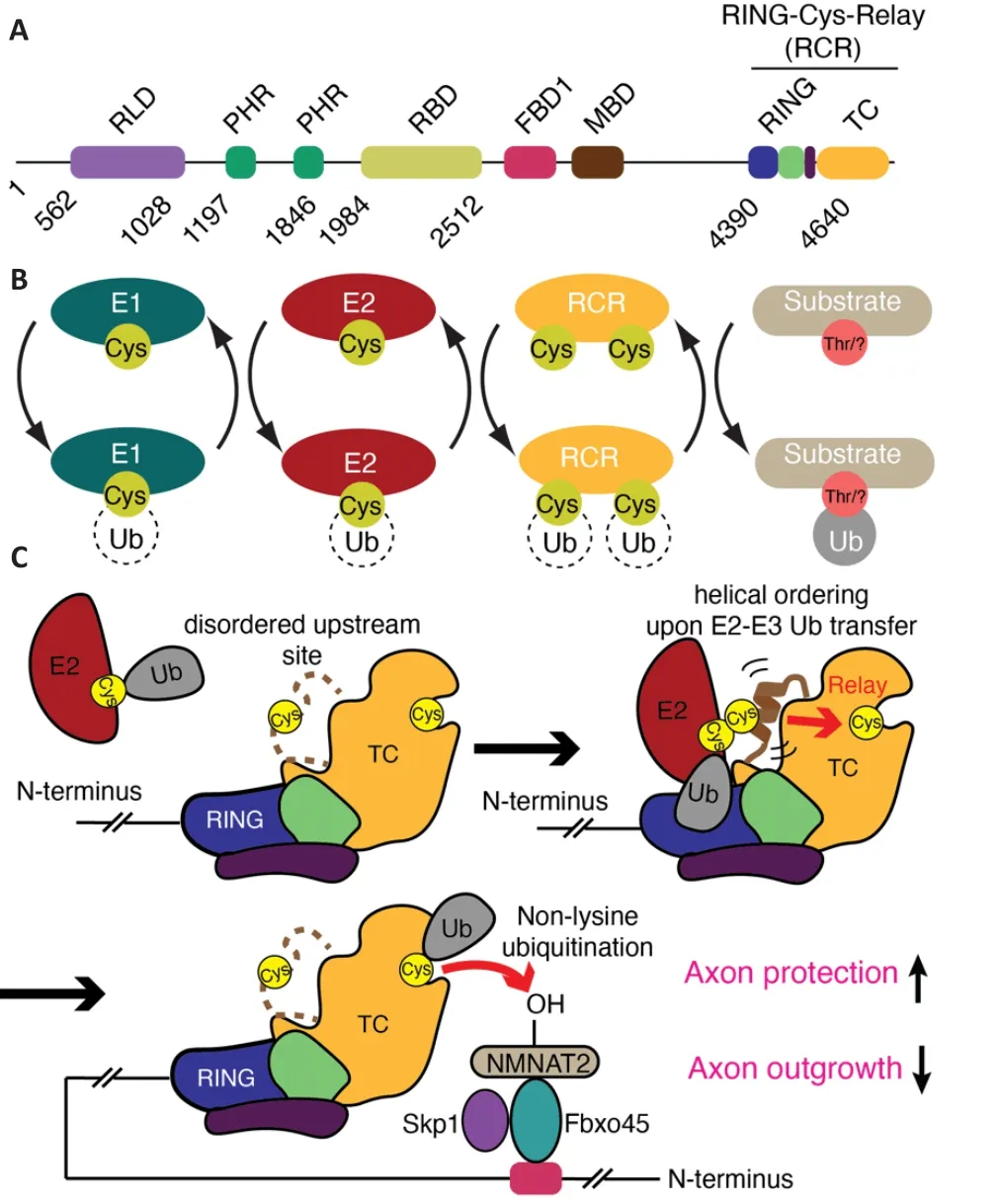
Figure 2 | The mechanism of MYCBP2 RING-Cys-Relay activity and its role in axon protection and neural development.
RING-Cys-Relay Ubiquitin Ligase Activity Is Required for Normal Neural Development
Whilst MYCBP2 ligase activity had been implicated with axon degeneration by tuning NMNAT levels in Drosophila (Xiong et al., 2012), it remained unknown if MYCBP2’s newly discovered RCR mechanism was central to its pro-degenerative function.To test this, a CRISPR knock-in mouse model has been generated consisting of a constitutive mutation of the upstream cysteine to a catalytically dead alanine (Mabbitt et al., 2020).Whilst the full-length protein is preserved, ubiquitin transfer to the upstream cysteine, subsequent relay and ensuing ester-linked ubiquitination is ablated.A striking reduction in neurite outgrowth was observed in superior cervical ganglion explants from this homozygous knock-in model, reminiscent of the phenotypes observed with MYCBP2 silencing in mammals (Burgess et al., 2004; D’Souza et al., 2005;Bloom et al., 2007).Indeed, loss of RCR ubiquitin ligase activity in late-stage embryos also resulted in a marked reduction in diaphragm innervation by the phrenic nerve (Mabbitt et al., 2020).These observations indicated that the newly discovered RCR ubiquitin ligase mechanism is associated with the previously reported neurodevelopmental phenotypes.
RING-Cys-Relay Ubiquitin Ligase Activity Promotes Wallerian Degeneration
Notably, in addition to the neurodevelopmental phenotypes being recapitulated in the homozygous knock-in mouse model, so were the axonprotective effects.Despite the reduction in neurite outgrowth, loss of RCR ubiquitin ligase activity attenuated the degeneration of superior cervical ganglia explants following axotomy (Mabbitt et al., 2020).In addition,exogenous NMNAT2 was stabilized in embryonic fibroblasts from homozygous and heterozygous knock-in mice.Conditional silencing of MYCBP2 has also been shown to preserve dorsal root ganglia neurons following axotomy and vincristine treatment (Babetto et al., 2013).Whether protection is conferred after vincristine treatment in RCR-defective mice remains to be tested.
An exciting prospect is the stunted growth phenotype in the constitutive knock-in mouse model suppresses the potency of the protective effect after axotomy rather than exacerbates it.Hence, protection might be more pronounced if loss of RCR activity were to be induced pharmacologically in a post-developmental context.However, it remains unclear whether the demonstrated role of MYCBP2 RCR activity in neurodevelopment is amenable to sustained suppression in adult animals without significant side effects.Generation of a conditional knock-in mouse model, or tool inhibitors,should allow these aspects to be investigated further.Taken together, these observations suggest that the neurodevelopmental and pro-degenerative effects of MYCBP2 are largely dependent on its unusual RCR E3 activity.
Structural and Biochemical Insights into RING-Cys-Relay Ubiquitin Ligase Activity
Enabling the discovery of small molecule inhibitors of the RCR ubiquitin ligase machinery in MYCBP2 would be its structural and biochemical characterization.Blocking the binding of the ubiquitin-loaded E2 enzyme,impairing the ubiquitin relay process, or blocking the downstream site would be expected to abolish MYCBP2 ubiquitin ligase activity.Structural studies initially revealed the general architecture of the RCR module and its downstream catalytic site (Pao et al., 2018).A fortuitous packing interaction propagated throughout the crystal placed the amino acid threonine, which has a hydroxyl side chain, within the downstream active site.This provided insights into how substrate selection mediated at the catalytic site is achieved and how it might be leveraged for inhibitor design (Pao et al., 2018).However,a number of mechanistic details remained poorly understood.For example,it was not known if the RING-finger domain binds E2 enzyme as observed for other RING domain-containing E3s.The upstream site was also found to be completely disordered in the initial isolated structure.Hence, it was also unclear how the upstream cysteine accepted ubiquitin from E2 enzyme and how the relay process would deliver the ubiquitin cargo to the downstream site.
By further leveraging the activity-based probe technology a stabilized form of the otherwise labile ternary E2-E3-ubiquitin transfer intermediate was prepared and isolated (Mabbitt et al., 2020).This permitted high-resolution structure determination by X-ray crystallography.The structure revealed the molecular contacts required for E2-E3 ubiquitin transfer.In the earlier isolated structure, the region containing the upstream cysteine, termed the mediator loop, was too flexible to be observed (Pao et al., 2018).The new structure of the stabilized transfer complex revealed that the region containing the upstream cysteine forms a transiently ordered helical configuration during E2-E3 ubiquitin transfer (Figure 2C).The study also provides insights into how the intramolecular ubiquitin relay step works.Proline scanning experiments supported a model where the energy required to facilitate the striking ubiquitin relay process is generated by the transient ordering of the upstream site.This might then act like an “entropic spring” enabling the ubiquitin molecule to be catapulted to the downstream site (Figure 2C) (Mabbitt et al.,2020).
Substrate Recognition Is Achieved through a Multi-Subunit MYBCP2 Complex
The described RCR ubiquitin ligase module resides at the extreme C-terminus of the giant MYCBP2 protein but a region in the middle of the protein,known as the Fsn-1/Fbxo45 binding domain 1 (FBD1) (Figure 2A), seemingly cooperates with the RCR module.In mammals the FBD1 region in MYCBP2 binds the F-box domain containing protein Fbxo45 and the adapter protein Skp1 and this subcomplex serves as a substrate receptor module (Figure 2C)(Liao et al., 2004; Wu et al., 2007; Saiga et al., 2009).In C.elegans the same interaction is found between the orthologous proteins (Rpm-1 and Fsn-1).The formation of multi-subunit ubiquitin ligase complexes, where the substrate receptor module is a distinct polypeptide, is the hallmark of a large subset of E3s known as the Cullin E3s (Baek et al., 2020).However, unlike Cullin E3s that dynamically exchange substrate receptor modules (Wang et al., 2020), Fbxo45 is the only receptor module assigned to MYCBP2.The fact that MYCBP2, a non-Cullin family member, also utilizes a dedicated substrate receptor is a further irregularity to its E3 mechanism.
The FBD1 region in MYCBP2 is ~2000 residues N-terminal of the RCR ubiquitin ligase module (Figure 2A) (Saiga et al., 2009; Desbois et al.,2018).Knock-down of Skp1 or Fbxo45 phenocopies MYCBP2 silencing as it confers axon-protection in response to both physical and chemical injury(Yamagishi and Tessier-Lavigne, 2016).Furthermore, this has been directly linked to stabilization of NMNAT2 (Yamagishi and Tessier-Lavigne, 2016).Consistent with the role of F-box domains being direct substrate receptors,Fbxo45 interacts with NMNAT2 (Babetto et al., 2013; Desbois et al., 2018).Interestingly, disruption of the interaction between Rpm-1 and Fsn-1 in C.elegans with a transgenically expressed peptide inhibitor recapitulates the synaptic defects observed with null worms (Sharma et al., 2014).This suggests that the substrate receptor sub-complex is also a potential therapeutic target.Taken together, these findings point toward a huge multi-subunit ligase machine, where the C-terminal RCR module is the catalytic engine, being central to normal neurodevelopment and post-developmental programmed axon degeneration (Figure 2C).Hence, inhibitors that disrupt not only the RCR module, but also the formation of the multi-subunit complex, should stabilize NMNAT2 and confer axon protective effects in response to injury.Further structural characterization should enable the visualization of this complex and demonstrate how Fbxo45 binds substrates and places them proximal to the C-terminal RCR ubiquitin ligase module, and also provide insights into how this activity is regulated.
Outlook
Beyond these findings, many questions remain unanswered.Ubiquitination of NMNAT2 on an amino acid with a hydroxyl side chain is most likely to be central to destabilizing NMNAT2 and promoting programmed axon degeneration.However, the high activity of MYCBP2 towards small molecule hydroxy compounds raises the possibility that a non-proteinaceous substrate might also be involved, as has now been observed with another unusual E3 ligase that has recently come to light (Otten et al., 2021).Furthermore,we do not appreciate the limitations of MYCBP2 and SARM1 inhibition.Thus, it remains possible these are complementary targets and inhibition of one rather than the other might be preferable for certain indications or neuron types.Although there are data in support of MYCBP2 operating upstream of SARM1 in a linear pathway, Drosophila genetics indicates that MYCBP2 might operate in parallel of SARM1 (Neukomm et al., 2017).It is also important to consider the roles MYCBP2 E3 ligase activity has outside neurodevelopment and axon degeneration.Recently, Rpm-1 has been shown to suppress neuronal autophagy by destabilizing the autophagy initiator UNC51/ULK1 (Crawley et al., 2019).Whether this relates to the established neurological phenotypes and broadens or limits the therapeutic scope of MYCBP2 inhibition remains unclear.Interestingly, MYCBP2 links to cancer have also been reported as it destabilizes the tumor suppressor FBW7 thereby contributing to chemotherapy resistance (Richter et al., 2020).In addition,MYCBP2 can regulate cell growth and proliferation via the mTOR pathway(Murthy et al., 2004; Han et al., 2008, 2012).
The role of the ubiquitin system in WD does not end with MYCBP2.Forward genetic screens in Drosophila have also identified a third pro-degenerative protein (Neukomm et al., 2017).Axundead belongs to the kelch-like gene family and Axundead mutants suppress axon death across multiple neuron types and preserve axon morphology for the lifespan of the animal (Neukomm et al., 2017).Conceptually similar to Fbxo45, kelch-like proteins are the the substrate receptors for the multi-subunit Cullin E3s (Shi et al., 2019).Although conservation of this pathway in mammals remains to be confirmed,it might also afford an additional opportunity for therapeutic intervention.Furthermore, NAD+and nicotinamide mononucleotide levels have been shown to regulate WD (Di Stefano et al., 2015; Figley et al., 2021).Strategies that modulate the axonal levels of these nucleotides might also halt or retard axonal degeneration.For example, activators of NMNAT2 would be expected to enhance NAD+ levels and inhibit WD.In summary a deeper molecular level understanding of multiple regulators of programmed axon degeneration has emerged.These might now be exploited and bring us ever closer to much needed therapeutic relief against chemotherapy-induced neuropathies,neuronal injury and neurodegenerative disease.
Acknowledgments:The author would like to thank Dr.Marc-Andre Dery for help drafting the manuscript.
Author contributions:The author completed the manuscript independently and approved the final manuscript.
Conflicts of interest:SV is founder and consultant of Outrun Therapeutics, a biotechnology company working in the ubiquitin field.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Interplay of SOX transcription factors and microRNAs in the brain under physiological and pathological conditions
- Cerebellar pathology in motor neuron disease:neuroplasticity and neurodegeneration
- Neuroinflammation as a mechanism linking hypertension with the increased risk of Alzheimer’s disease
- The endogenous progenitor response following traumatic brain injury: a target for cell therapy paradigms
- The relationship between amyloid-beta and brain capillary endothelial cells in Alzheimer’s disease
- Telomerase and neurons: an unusual relationship